来源于光黑壳属菌株的抗病毒化合物及其制备方法与应用
1.本发明属于生物医药领域,涉及一种来源于光黑壳属菌株的抗病毒化合物及其制备方法与应用。
背景技术:
2.流行性感冒简称流感,是由流感病毒引起的急性发热性呼吸道传染病,具有流行面广、传染性强、发病率高等特点。流感病毒是负链rna病毒,属于正黏病毒科家族。根据其基质蛋白和核蛋白的不同,可分为甲型流感病毒和乙型流感病毒和丙型流感病毒。其中甲型流感病毒(influenza a virus,iav)具有更强的抗原变异性,对人类健康更具严重威胁。临床上预防和治疗流感的手段主要有疫苗和抗流感药物,但是由于流感病毒具有极强的重组变异和抗原漂移能力,严重地限制了流感疫苗的免疫保护作用。同时,目前的流感病毒流行株对临床一线抗流感药物也产生了不同程度的耐受,因此亟需发展新型抗流感病毒药物。
3.寨卡病毒(zika virus,zikv)属于蚊媒传播的黄病毒属病毒,除以伊蚊为传播媒介外,同时还有垂直传播、血液传播、飞沫传播、母乳传播、性传播等途径。其基因组为单股正链rna,全长约11kb,编码一个开放读码框(open reading frame,orf),可形成一个多聚蛋白。随着不断变异,寨卡病毒传染力和致病力越来越强。近几十年来,人们一直致力于抗寨卡病毒药物的研发,但尚未有该类药物和疫苗上市。因此,快速发现,准确捕获对寨卡病毒具有抑制作用的分子,尤其新结构新机制小分子,为其药物研发提供前期储备,具有重大的战略意义。
技术实现要素:
4.本发明的目的是提供一种来源于光黑壳属菌株的抗病毒化合物及其制备方法与应用。
5.本发明提供了化合物或其药学上可接受的盐在制备病毒抑制剂或抗病毒药物中的应用。
6.本发明还保护化合物。
7.本发明还保护化合物的制备方法,包括如下步骤:发酵培养光黑壳属菌株cpcc 400972,得到化合物。
8.本发明还保护光黑壳属菌株cpcc 400972。
9.本发明还保护光黑壳属菌株cpcc 400972的培养物,是将光黑壳属菌株cpcc 400972进行培养得到的物质。
10.本发明还保护光黑壳属菌株cpcc 400972在制备化合物中的应用。
11.本发明还保护一种病毒抑制剂或抗病毒药物,其含有化合物或其药学上可接受的盐。
12.本发明还保护光黑壳属菌株cpcc 400972或/和所述培养物在制备病毒抑制剂或
抗病毒药物中的应用。
13.本发明还保护病毒抑制剂或抗病毒药物,其含有光黑壳属菌株cpcc 400972或/和所述培养物。
14.本发明还保护病毒抑制剂或抗病毒药物的制备方法,包括如下步骤:将光黑壳属菌株cpcc400972或/和所述培养物作为病毒抑制剂或抗病毒药物的成分,得到所述病毒抑制剂或抗病毒药物。
15.光黑壳属菌株cpcc 400972全称为光黑壳属菌株(preussia sp.)cpcc 400972,已于 2020年11月26日保藏于中国微生物菌种保藏管理委员会普通微生物中心(简称cgmcc,地址为:北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所),保藏登记号为cgmccno.21069。
16.示例性的,发酵培养光黑壳属菌株cpcc 400972的方法包括如下步骤:将光黑壳属菌株 cpcc 400972在发酵培养基中培养(具体可为静置培养),得到发酵产物。
17.示例性的,将光黑壳属菌株cpcc 400972进行培养的方法包括如下步骤:将光黑壳属菌株cpcc 400972在发酵培养基中培养(具体可为静置培养),得到发酵产物。
18.示例性的发酵培养基的原料组成:100g大米和100ml水。
19.示例性的发酵培养基的制备方法:100g大米和100ml水,浸泡(28℃,24小时)。
20.化合物的制备方法中还包括纯化步骤。
21.所述纯化步骤包括提取。
22.所述提取具体为采用甲醇提取。
23.所述纯化步骤包括萃取。
24.所述萃取具体为采用乙酸乙酯萃取。
25.所述纯化步骤具体见实施例2的步骤二。
26.化合物的制备方法中还包括化学合成的步骤。
27.所述化学合成步骤具体见实施例3。
28.以上任一所述病毒可为rna病毒。
29.以上任一所述病毒可为单链rna病毒。
30.以上任一所述病毒可为黄病毒科病毒或正黏液病毒科病毒。
31.所述黄病毒科病毒具体可为黄病毒属病毒。所述黄病毒属病毒具体可为寨卡病毒。
32.以上任一所述正黏液病毒科病毒可为流感属病毒。所述流感属病毒具体可为流感病毒。所述流感病毒具体可为甲型流感病毒。所述流感病毒具体可为h1n1亚型流感病毒。
33.以上任一所述化合物为式(ⅰ)所示化合物或式(ⅱ)所示化合物;
[0034][0035]
式(ⅰ)中,r1为羧基或酯基或烷基;
[0036]
式(ⅰ)中,r2为烷基;
[0037]
式(ⅰ)中,r3为
[0038]
式(ⅰ)中,r4为
[0039]
ra为rb为rc为x为o或n;
[0040][0041]
式(ⅱ)中,r1为
[0042]
式(ⅰ)中,r1具体可为具体可为或异丁烯基或异丁烯酸基或异丁烯醇基等。
[0043]
式(ⅰ)中,r2具体可为甲基或乙基或丙基或丁基等。
[0044]
所述化合物具体为化合物1至化合物27中的任意一种;
[0045][0046]
上述药物中,化合物或化合物药学上可接受的盐可作为有效成分之一,也可作为
唯一有效成分。上述药物中,化合物或化合物药学上可接受的盐可作为活性成分之一,也可作为唯一活性成分。
[0047]
上述药物中,还含有载体材料。载体材料包括但不限于水溶性载体材料(如聚乙二醇、聚乙烯吡咯烷酮、有机酸等)、难溶性载体材料(如乙基纤维素、胆固醇硬脂酸酯等)、肠溶性载体材料(如醋酸纤维素酞酸酯和羧甲乙纤维素等)。使用这些材料可以制成多种剂型,包括但不限于片剂、胶囊、滴丸、气雾剂、丸剂、粉剂、溶液剂、混悬剂、乳剂、颗粒剂、脂质体、透皮剂、口含片、栓剂、冻干粉针剂等。可以是普通制剂、缓释制剂、控释制剂及各种微粒给药系统。为了将单位给药剂型制成片剂,可以广泛使用本领域公知的各种载体。关于载体的例子是,例如稀释剂与吸收剂,如淀粉、糊精、硫酸钙、乳糖、甘露醇、蔗糖、氯化钠、葡萄糖、尿素、碳酸钙、白陶土、微晶纤维素、硅酸铝等;湿润剂与粘合剂,如水、甘油、聚乙二醇、乙醇、丙醇、淀粉浆、糊精、糖浆、蜂蜜、葡萄糖溶液、阿拉伯胶浆、明胶浆、羧甲基纤维素钠、紫胶、甲基纤维素、磷酸钾、聚乙烯吡咯烷酮等;崩解剂,例如干燥淀粉、海藻酸盐、琼脂粉、褐藻淀粉、碳酸氢钠与枸橼酸、碳酸钙、聚氧乙烯、山梨糖醇脂肪酸酯、十二烷基磺酸钠、甲基纤维素、乙基纤维素等;崩解抑制剂,例如蔗糖、三硬脂酸甘油酯、可可脂、氢化油等;吸收促进剂,例如季铵盐、十二烷基硫酸钠等;润滑剂,例如滑石粉、二氧化硅、玉米淀粉、硬脂酸盐、硼酸、液体石蜡、聚乙二醇等。还可以将片剂进一步制成包衣片,例如糖包衣片、薄膜包衣片、肠溶包衣片,或双层片和多层片。为了将单位给药剂型制成丸剂,可以广泛使用本领域公知的各种载体。关于载体的例子是,例如稀释剂与吸收剂,如葡萄糖、乳糖、淀粉、可可脂、氢化植物油、聚乙烯吡咯烷酮、高岭土、滑石粉等;粘合剂如阿拉伯胶、黄蓍胶、明胶、乙醇、蜂蜜、液糖、米糊或面糊等;崩解剂,如琼脂粉、干燥淀粉、海藻酸盐、十二烷基磺酸钠、甲基纤维素、乙基纤维素等。为了将单位给药剂型制成栓剂,可以广泛使用本领域公知的各种载体。关于载体的例子是,例如聚乙二醇、卵磷脂、可可脂、高级醇、高级醇的酯、明胶、半合成甘油酯等。为了将单位给药剂型制成注射用制剂,如溶液剂、乳剂、冻干粉针剂和混悬剂,可以使用本领域常用的所有稀释剂,例如,水、乙醇、聚乙二醇、1,3-丙二醇、乙氧基化的异硬脂醇、多氧化的异硬脂醇、聚氧乙烯山梨醇脂肪酸酯等。另外,为了制备等渗注射液,可以向注射用制剂中添加适量的氯化钠、葡萄糖或甘油,此外,还可以添加常规的助溶剂、缓冲剂、ph调节剂等。此外,如需要,也可以向药物制剂中添加着色剂、防腐剂、香料、矫味剂、甜味剂或其它材料。使用上述剂型可以经注射给药,包括皮下注射、静脉注射、肌肉注射和腔内注射等;腔道给药,如经直肠和阴道;呼吸道给药,如经鼻腔;粘膜给药。
[0048]
本发明对于病毒防控和病毒引起的疾病的防控具有重大的应用推广价值。
附图说明
[0049]
图1为化合物1的1h-nmr谱。
[0050]
图2为化合物2的1h-nmr谱。
[0051]
图3为化合物3的1h-nmr谱。
[0052]
图4为化合物4的1h-nmr谱。
[0053]
图5为化合物5的1h-nmr谱。
[0054]
图6为化合物6的1h-nmr谱。
[0055]
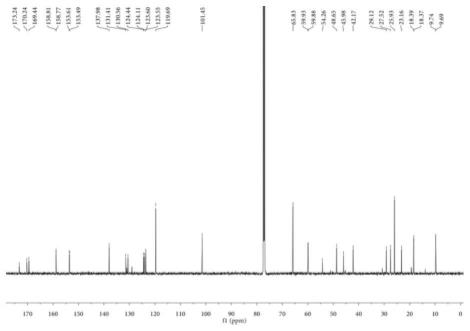
图7为化合物7的1h-nmr谱。
[0056]
图8为化合物8的1h-nmr谱。
[0057]
图9为化合物9的1h-nmr谱。
[0058]
图10为化合物10的1h-nmr谱。
[0059]
图11为化合物11的1h-nmr谱。
[0060]
图12为化合物12的1h-nmr谱。
[0061]
图13为化合物13的1h-nmr谱。
[0062]
图14为化合物14的1h-nmr谱。
[0063]
图15为化合物15的1h-nmr谱。
[0064]
图16为化合物16的h-nmr谱。
[0065]
图17为化合物17的h-nmr谱。
[0066]
图18为化合物18的h-nmr谱。
[0067]
图19为化合物19的h-nmr谱。
[0068]
图20为化合物20的h-nmr谱。
[0069]
图21为化合物21的h-nmr谱。
[0070]
图22为化合物22的1h-nmr谱。
[0071]
图23为化合物23的1h-nmr谱。
[0072]
图24为化合物24的1h-nmr谱。
[0073]
图25为化合物25的1h-nmr谱。
[0074]
图26为化合物26的1h-nmr谱。
[0075]
图27为化合物27的1h-nmr谱。
[0076]
图28为化合物1的
13
c-nmr谱。
[0077]
图29为化合物2的
13
c-nmr谱。
[0078]
图30为化合物3的
13
c-nmr谱。
[0079]
图31为化合物4的
13
c-nmr谱。
[0080]
图32为化合物5的
13
c-nmr谱。
[0081]
图33为化合物6的
13
c-nmr谱。
[0082]
图34为化合物7的
13
c-nmr谱。
[0083]
图35为化合物8的
13
c-nmr谱。
[0084]
图36为化合物9的
13
c-nmr谱。
[0085]
图37为化合物10的
13
c-nmr谱。
[0086]
图38为化合物11的
13
c-nmr谱。
[0087]
图39为化合物12的
13
c-nmr谱。
[0088]
图40为化合物13的
13
c-nmr谱。
[0089]
图41为化合物14的
13
c-nmr谱。
[0090]
图42为化合物15的
13
c-nmr谱。
[0091]
图43为化合物16的
13
c-nmr谱。
[0092]
图44为化合物17的
13
c-nmr谱。
[0093]
图45为化合物18的
13
c-nmr谱。
[0094]
图46为化合物19的
13
c-nmr谱。
[0095]
图47为化合物20的
13
c-nmr谱。
[0096]
图48为化合物21的
13
c-nmr谱。
[0097]
图49为化合物22的
13
c-nmr谱。
[0098]
图50为化合物23的
13
c-nmr谱。
[0099]
图51为化合物24的
13
c-nmr谱。
[0100]
图52为化合物25的
13
c-nmr谱。
[0101]
图53为化合物26的
13
c-nmr谱。
[0102]
图54为化合物27的
13
c-nmr谱。
具体实施方式
[0103]
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进的指南,并不以任何方式构成对本发明的限制。下述实施例中的实验方法,如无特殊说明,均为常规方法,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。如无特殊说明,以下实施例中的定量试验,均设置三次重复实验,结果取平均值。
[0104]
实施例1、菌株的获得、鉴定和保藏
[0105]
一、菌株的获得
[0106]
从新疆自治区伊犁自治州巩留县采集紫毛蕊花(verbascumphoeniceuml.)植株。取整个植株,依次用75%酒精漂洗1min、2%次氯酸钠漂洗3min、75%酒精漂洗30s、无菌水漂洗3次,然后用无菌吸水纸将植株表面水吸干,并用无菌剪刀将植株剪成长约0.5cm的组织块,置于含有硫酸链霉素(50mg l-1
)和四环素(50mg l-1
)的pda固体培养基平板。将平板放置于28℃培养箱中培养5d-7d。根据菌落的颜色、大小、形状等特征挑取单菌落,置于含有pda培养基的试管斜面上,获得纯培养的菌株,待长好后,置于冰箱中保存。保存方法:于20%甘油冻存管-80℃保存藏。获得的一株菌命名为菌株cpcc 400972。
[0107]
二、菌株的鉴定
[0108]
菌株cpcc 400972的形态特征:菌落较大,菌落质地疏松,外观干燥,不透明,呈现或紧或松的蛛网状、绒毛状或棉絮状;菌落与培养基的连接紧密,不易挑取。菌株cpcc 400972 的分子鉴定:收集菌体,提取总dna,然后扩增its基因并测序,将测序结果在ncbi进行比对;its基因序列如序列表中序列1所示,与菌株preussia africana isolate a54(genbankno.kx611037.1)在its基因序列相似性达99.77%。
[0109]
根据形态特征和分子鉴定结果,菌株cpcc 400972属于光黑壳属菌株(preussia sp.),因此命名为光黑壳属菌株(preussia sp.)cpcc 400972。
[0110]
三、菌株的保藏
[0111]
光黑壳属菌株(preussia sp.)cpcc 400972,已于2020年11月26日保藏于中国微生物菌种保藏管理委员会普通微生物中心(简称cgmcc,地址为:北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所),保藏登记号为cgmcc no.21069。光黑壳属菌株(preussiasp.)cpcc 400972cgmcc no.21069简称为光黑壳属菌株cpcc 400972。
[0112]
实施例2、发酵光黑壳属菌株cpcc 400972制备化合物
[0113]
一、培养与发酵
[0114]
1、将光黑壳属菌株cpcc 400972孢子接种于斜面培养基,28℃培养5天,得到斜面菌种。
[0115]
斜面培养基由马铃薯浸粉、葡萄糖、琼脂和水组成。各成分在斜面培养基中浓度分别为:马铃薯浸粉4g/l,葡萄糖20g/l,琼脂15g/l。所述斜面培养基的ph值为5.6
±
0.2。
[0116]
2、从斜面菌种挑取菌丝体,接种至装有100ml种子培养基的500ml玻璃瓶中,28℃、 200rpm振荡培养2天,得到种子液。
[0117]
种子培养基由马铃薯浸粉、葡萄糖和水组成。各成分在种子培养基中浓度分别为:马铃薯浸粉4g/l,葡萄糖20g/l。所述种子培养基的ph值为5.6
±
0.2。
[0118]
3、将10ml种子液接种至装有发酵培养基的三角瓶中,28-30℃静置培养40天(培养 7天后在无菌环境下将培养基打散至小块,培养期间可不定期摇晃,以保证培养过程中空气的进入),得到发酵产物(即发酵培养的整个体系)。
[0119]
装有发酵培养基的三角瓶:取500ml三角瓶,加入100g大米和100ml水,浸泡(28℃,24小时),然后121℃灭菌20分钟。
[0120]
进行多个重复处理,获得4000g发酵产物。
[0121]
二、从发酵产物中分离纯化化合物
[0122]
1、取4000g步骤一的发酵产物,使用甲醇超声提取三次(具体步骤:加入5l甲醇,室温环境下,50khz超声提取30分钟,收集有机相;剩余水相再加入5l甲醇,室温环境下,50khz超声提取30分钟,收集有机相;剩余水相再加入5l甲醇,室温环境下,50khz 超声提取30分钟,收集有机相;将三次的有机相合并),然后采用孔径3mm的布氏漏斗过滤,收集滤液(约15l),进行减压浓缩(75mbar)以去除溶剂,得到甲醇粗提物(约100g)。
[0123]
2、取步骤1获得的全部甲醇粗提物,用1l水分散,然后用乙酸乙酯萃取三次(具体步骤:加入1l乙酸乙酯进行萃取,收集有机相;剩余水相再加入1l乙酸乙酯进行萃取,收集有机相;剩余水相再加入1l乙酸乙酯进行萃取,收集有机相;将三次的有机相合并),然后进行减压浓缩(75mbar)以去除溶剂,得到乙酸乙酯提取物(32.2g)。
[0124]
3、取步骤2获得的全部乙酸乙酯提取物,溶解于20ml甲醇,然后加入35g硅胶(200-300 目),均匀拌样,然后进行硅胶柱层析分离。
[0125]
硅胶柱层析的参数如下:
[0126]
柱子型号:6.5cm
×
60cm;填充介质:200-300目硅胶(青岛海洋),400g;
[0127]
洗脱程序如下:
①
先用二氯甲烷洗脱3000ml;
②
接着用98:2的二氯甲烷-甲醇溶液(由98体积份二氯甲烷和2体积份甲醇组成的溶液)洗脱5000ml;
③
接着用96:4的二氯甲烷
‑ꢀ
甲醇溶液(由96体积份二氯甲烷和4体积份甲醇组成的溶液)洗脱5000ml;
④
接着用94:6 的二氯甲烷-甲醇溶液(由96体积份二氯甲烷和4体积份甲醇组成的溶液)洗脱5000ml;
⑤
接着用90:10的二氯甲烷-甲醇溶液(由90体积份二氯甲烷和10体积份甲醇组成的溶液)洗脱5000ml;
⑥
接着用80:20的二氯甲烷-甲醇溶液(由80体积份二氯甲烷和20体积份甲醇组成的溶液)洗脱3000ml;
⑦
接着用甲醇洗脱4000ml。从洗脱程序开始不间断收集洗脱出的液体,每份收集500ml,共收集60份,依次命名为馏分1至馏分60。经过tlc检测,将馏分合并然后减压浓缩去除溶剂,具体如下:馏分1-5合并,然后减压浓缩去除溶剂,得到馏分b1(2.6g);馏分6-15合并,然后减压浓缩去除溶剂,得到馏分b2(4.1g);馏分16-25 合并,然后
减压浓缩去除溶剂,得到馏分b3(3.2g);馏分26-35合并,然后减压浓缩去除溶剂,得到馏分b4(7.2g);馏分36-40合并,然后减压浓缩去除溶剂,得到馏分b5(18.8 g);馏分41-50合并,然后减压浓缩去除溶剂,得到馏分b6(0.5g);馏分51-55合并,然后减压浓缩去除溶剂,得到馏分b7(0.8g);馏分56-60合并,然后减压浓缩去除溶剂,得到馏分b8(1.5g)。
[0128]
4、取步骤3收集的馏分b4(7.2g),溶解于10ml甲醇,加入7.2g硅胶(200-300目),均匀拌样,然后进行加压硅胶柱层析分离。
[0129]
加压硅胶柱层析的参数如下:
[0130]
柱子型号:6cm
×
25cm;填充介质:200-300目硅胶(青岛海洋),140g;
[0131]
压力:100mbar;
[0132]
洗脱程序如下:用二氯甲烷-丙酮溶液(由10体积份二氯甲烷和1体积份丙酮组成的溶液)洗脱2000ml。从洗脱程序开始不间断收集洗脱出的液体,每份收集100ml,共收20份,依次命名为馏分1至馏分20。经过tlc检测,将馏分合并然后减压浓缩去除溶剂,具体如下:馏分1-3合并,然后减压浓缩去除溶剂,得到馏分c1(0.5g);馏分4-6合并,然后减压浓缩去除溶剂,得到馏分c2(1.0g);馏分7-9合并,然后减压浓缩去除溶剂,得到馏分c3 (0.6g);馏分10-13合并,然后减压浓缩去除溶剂,得到馏分c4(4.3g);馏分14-17合并,然后减压浓缩去除溶剂,得到馏分c5(1.0g);馏分18-20合并,然后减压浓缩去除溶剂,得到馏分c6(0.5g)。
[0133]
5、取馏分c4(4.3g),溶解于5ml甲醇,加入4.3g mc ods-aq-hg填料拌匀,得到固体样品;将固体样品填充至样品柱,然后连接色谱柱,用色谱仪进行分离纯化。
[0134]
色谱柱:flash spherical c18 20-35μm 100a,80g。
[0135]
色谱仪:美国teledyne isco公司combiflash rf 200制备型色谱仪。
[0136]
流动相a:由三氟乙酸和水组成,三氟乙酸浓度为0.01%(体积比)。流动相b:由三氟乙酸和乙腈组成,三氟乙酸浓度为0.01%(体积比)。流动相流速:30ml/min。
[0137]
检测波长:210nm。
[0138]
洗脱程序:0-60min,流动相b占流动相的体积分数由10%线性上升至100%,相应的流动相a占流动相的体积分数由90%线性下降至0%;60-70min,流动相为流动相b。
[0139]
整个洗脱过程共获得4个馏分(每个馏分即对应一个洗脱峰的过柱后溶液,4个馏分依次命名为馏分d1-d4)。馏分d1对应保留时间tr为5-20min的洗脱峰,馏分d2对应保留时间tr为20-30min的洗脱峰,馏分d3对应保留时间tr为30-32min的洗脱峰,馏分d4对应保留时间tr为40-50min的洗脱峰。
[0140]
馏分d3,减压浓缩以去除溶剂,称重为1.03g,即为化合物1。
[0141]
馏分d1,减压浓缩以去除溶剂,溶解于3ml甲醇,即为馏分d1的浓缩液。
[0142]
馏分d2,减压浓缩以去除溶剂,溶解于2ml甲醇,即为馏分d2的浓缩液。
[0143]
馏分d4,减压浓缩以去除溶剂,溶解于2ml甲醇,即为馏分d4的浓缩液。
[0144]
6、取3ml馏分d1的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0145]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0146]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0147]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 20%(体积比)。流动相流速为4ml/min。
[0148]
检测波长:210nm。
[0149]
收集峰值对应的保留时间tr为14.5min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物15(2.3mg)。
[0150]
收集峰值对应的保留时间tr为21.8min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物16(101.2mg)。
[0151]
7、取2ml馏分d2的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0152]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0153]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0154]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 35%(体积比)。流动相流速为4ml/min。
[0155]
检测波长:210nm。
[0156]
收集峰值对应的保留时间tr为29.1min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物17(5.2mg)。
[0157]
收集峰值对应的保留时间tr为21.8min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物2(8.2mg)。
[0158]
8、取2ml馏分d4的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0159]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0160]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0161]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为50%(体积比)。流动相流速为2.5ml/min。
[0162]
检测波长:210nm。
[0163]
收集峰值对应的保留时间tr为19.6min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物22(11.4mg)。
[0164]
9、取步骤3收集的馏分b5(18.8g),用甲醇溶解,加入18.8g硅胶(200-300目)拌匀,进行加压硅胶柱层析分离。
[0165]
加压硅胶柱层析的参数如下:
[0166]
柱子型号:6cm
×
25cm;填充介质:200-300目硅胶(青岛海洋),140g;
[0167]
压力:100mbar;
[0168]
流动相a为二氯甲烷。流动相b为丙酮。流动相流速:30ml/min。
[0169]
洗脱程序:0-100min,流动相b占流动相的体积分数由9%线性上升至100%,相应的流动相a占流动相的体积分数由91%线性下降至0%。从洗脱程序开始不间断收集洗脱出的液体,每份收集150ml,共收20份,依次命名为馏分1至馏分20。经过tlc检测,将馏分合并然后减压浓缩去除溶剂,具体如下:馏分1-4合并,然后减压浓缩去除溶剂,得到馏分c7(0.7g);馏分5-10合并,然后减压浓缩去除溶剂,得到馏分c8(3.1g);馏分11-15合并,然后减压浓缩去除溶剂,得到馏分c9(1.9g);馏分16-18合并,然后减压浓缩去除溶剂,得到馏分c10(5.5g);馏分19-20合并,然后减压浓缩去除溶剂,得到馏分c11(2.6g)。
[0170]
10、取馏分c8(3.1g),溶解于5ml甲醇,加入3.1g ymc ods-aq-hg填料拌匀,得到固体样品;将固体样品填充至样品柱,然后连接色谱柱,用色谱仪进行分离纯化。
[0171]
色谱柱:flash spherical c18 20-35μm 100a,80g。
[0172]
色谱仪:美国teledyne isco公司combiflash rf 200制备型色谱仪。
[0173]
流动相a:由三氟乙酸和水组成,三氟乙酸浓度为0.01%(体积比)。流动相b:由三氟乙酸和乙腈组成,三氟乙酸浓度为0.01%(体积比)。流动相流速:30ml/min。
[0174]
检测波长:210nm。
[0175]
洗脱程序:0-70min,流动相b占流动相的体积分数由10%线性上升至100%,相应的流动相a占流动相的体积分数由90%线性下降至0%;70-90min,流动相为流动相b。
[0176]
整个洗脱过程共获得4个馏分(每个流份即对应一个洗脱峰的过柱后溶液,4个馏分依次命名为馏分d5-d8)。馏分d5对应保留时间tr为20-30min的洗脱峰,馏分d6对应保留时间tr为5-15min的洗脱峰,馏分d7对应保留时间tr为35-40min的洗脱峰,馏分d8对应保留时间tr为40-50min的洗脱峰。
[0177]
馏分d5,减压浓缩以去除溶剂,溶解于2.5ml甲醇,即为馏分d5的浓缩液。
[0178]
馏分d6,减压浓缩以去除溶剂,溶解于2.5ml甲醇,即为馏分d6的浓缩液。
[0179]
馏分d7,减压浓缩以去除溶剂,溶解于1.5ml甲醇,即为馏分d7的浓缩液。
[0180]
馏分d8,减压浓缩以去除溶剂,溶解于1.5ml甲醇,即为馏分d8的浓缩液。
[0181]
11、取2.5ml馏分d5的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0182]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0183]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0184]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 25%(体积比)。流动相流速为2.5ml/min。
[0185]
检测波长:210nm。
[0186]
收集峰值对应的保留时间tr为29.7min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物10(4.5mg)。
[0187]
收集峰值对应的保留时间tr为36.0min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物19(11.8mg)。
[0188]
收集峰值对应的保留时间tr为42.9min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物11(2.7mg)。
[0189]
收集峰值对应的保留时间tr为43.1min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物20(23.2mg)。
[0190]
12、取2.5ml馏分d6的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0191]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0192]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0193]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 18%(体积比)。流动相流速为2.5ml/min。
[0194]
检测波长:210nm。
[0195]
收集峰值对应的保留时间tr为23.3min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物12(12.9mg)。
[0196]
13、取1.5ml馏分d7的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0197]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0198]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0199]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 45%(体积比)。流动相流速为2.5ml/min。
[0200]
检测波长:210nm。
[0201]
收集峰值对应的保留时间tr为18.9min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物18(21.3mg)。
[0202]
14、取1.5ml馏分d8的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0203]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0204]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0205]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 48%(体积比)。流动相流速为2.5ml/min。
[0206]
检测波长:210nm。
[0207]
收集峰值对应的保留时间tr为24.9min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物23(4.2mg)。
[0208]
15、取馏分c9(1.9g),溶解于3ml甲醇,加入1.9g ymc ods-aq-hg填料拌匀,得到固体样品;将固体样品填充至样品柱,然后连接色谱柱,用色谱仪进行分离纯化。
[0209]
色谱柱:flash spherical c18 20-35μm 100a,80g。
[0210]
色谱仪:美国teledyne isco公司combiflash rf 200制备型色谱仪。
[0211]
流动相a:由三氟乙酸和水组成,三氟乙酸浓度为0.01%(体积比)。流动相b:由三氟乙酸和乙腈组成,三氟乙酸浓度为0.01%(体积比)。流动相流速:20ml/min。
[0212]
检测波长:210nm。
[0213]
洗脱程序:0-70min,流动相b占流动相的体积分数由10%线性上升至100%,相应的流动相a占流动相的体积分数由90%线性下降至0%;70-90min,流动相为流动相b。
[0214]
整个洗脱过程共获得2个馏分(每个流份即对应一个洗脱峰的过柱后溶液,2个馏分依次命名为馏分d11-d12)。馏分d11对应保留时间tr为50-60min的洗脱峰,馏分d12对应保留时间tr为60-70min的洗脱峰。
[0215]
馏分d11,减压浓缩以去除溶剂,溶解于1.5ml甲醇,即为馏分d11的浓缩液。
[0216]
馏分d12,减压浓缩以去除溶剂,溶解于1.0ml甲醇,即为馏分d12的浓缩液。
[0217]
采用旋转蒸发仪分别浓缩蒸干各个流份,分别用适量甲醇溶解,即为各个流份的浓缩液。
[0218]
16、取1.5ml馏分d11的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0219]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0220]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0221]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 65%(体积比)。流动相流速为2.5ml/min。
[0222]
检测波长:210nm。
[0223]
收集峰值对应的保留时间tr为23.5min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物26(6.4mg)。
[0224]
17、取1.0ml馏分d12的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0225]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0226]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0227]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 90%(体积比)。流动相流速为2.5ml/min。
[0228]
检测波长:210nm。
[0229]
收集峰值对应的保留时间tr为26.8min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物27(8.7mg)。
[0230]
18、取馏分c10(5.5g),溶解于7ml甲醇,加入5.5g ymc ods-aq-hg填料拌匀,得到固体样品;将固体样品填充至样品柱,然后连接色谱柱,用色谱仪进行分离纯化。
[0231]
色谱柱:flash spherical c18 20-35μm 100a,80g。
[0232]
色谱仪:美国teledyne isco公司combiflash rf 200制备型色谱仪。
[0233]
流动相a:由三氟乙酸和水组成,三氟乙酸浓度为0.01%(体积比);流动相b:由乙腈组成,三氟乙酸浓度为0.01%(体积比)。流动相流速:30ml/min。
[0234]
检测波长:210nm。
[0235]
洗脱程序:0-70min,流动相b占流动相的体积分数由10%线性上升至100%,相应的流动相a占流动相的体积分数由90%线性下降至0%;70-90min,流动相为流动相b。
[0236]
整个洗脱过程共获得2个馏分(每个流份即对应一个洗脱峰的过柱后溶液,2个馏分依次命名为馏分d13-d14)。馏分d13对应保留时间20-35min的洗脱峰,馏分d14对应保留时间35-50min的洗脱峰。
[0237]
馏分d13,减压浓缩以去除溶剂,溶解于1.0ml甲醇,即为馏分d13的浓缩液。
[0238]
19、取1.0ml馏分d13的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0239]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0240]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0241]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 30%(体积比)。流动相流速为2.5ml/min。
[0242]
检测波长:210nm。
[0243]
收集峰值对应的保留时间tr为19.1min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物21(45.9mg)。
[0244]
20、取步骤3收集的馏分b3(3.2g),溶解于甲醇,加入3.2g硅胶(200-300目)拌匀,进行加压硅胶柱层析分离。
[0245]
加压硅胶柱层析的参数如下:
[0246]
柱子型号:4cm
×
25cm;填充介质:200-300目硅胶(青岛海洋),30g;
[0247]
压力:100mbar;
[0248]
流动相a为二氯甲烷。流动相b为丙酮。流动相流速为15ml/min。
[0249]
洗脱程序:0-100min,流动相a占流动相的体积分数由100%线性下降至91%,相应的流动相b占流动相的体积分数由0%线性上升至9%。从洗脱程序开始不间断收集洗脱出的液体,每份收集300ml,共收5份,依次命名为馏分1至馏分5。经过tlc检测,将馏分合并然后减压浓缩去除溶剂,具体如下:馏分1-3合并,然后减压浓缩去除溶剂,得到馏分c12(2.4g);馏分4-5合并,然后减压浓缩去除溶剂,得到馏分c13(1.7g)。
[0250]
21、取馏分c12(2.4g),溶解于3ml甲醇,加入2.4g ymc ods-aq-hg填料拌匀,得到
固体样品;将固体样品填充至样品柱,然后连接色谱柱,用色谱仪进行分离纯化。
[0251]
色谱柱:flash spherical c18 20-35μm 100a,80g。
[0252]
色谱仪:美国teledyne isco公司combiflash rf 200制备型色谱仪。
[0253]
流动相a:由三氟乙酸和水组成,三氟乙酸浓度为0.01%(体积比)。流动相b:由三氟乙酸和乙腈组成,三氟乙酸浓度为0.01%(体积比)。流动相流速:20ml/min。
[0254]
检测波长:210nm。
[0255]
洗脱程序:0-70min,流动相b占流动相的体积分数由10%线性上升至100%,相应的流动相a占流动相的体积分数由90%线性下降至0%;70-90min,流动相为流动相b。
[0256]
整个洗脱过程共获得2个馏分(每个流份即对应一个洗脱峰的过柱后溶液,2个馏分依次命名为馏分d9-d10)。馏分d9对应保留时间tr为50-60min的洗脱峰,馏分d10对应保留时间tr为60-70min的洗脱峰。
[0257]
馏分d9,减压浓缩以去除溶剂,溶解于1.0ml甲醇,即为馏分d9的浓缩液。
[0258]
馏分d10,减压浓缩以去除溶剂,溶解于1.0ml甲醇,即为馏分d10的浓缩液。
[0259]
22、取1.0ml馏分d9的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0260]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0261]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0262]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 80%(体积比)。流动相流速为2.5ml/min。
[0263]
检测波长:210nm。
[0264]
收集峰值对应的保留时间tr为15.4min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物5(7.5mg)。
[0265]
23、取1.0ml馏分d10的浓缩液,上样于色谱柱,用色谱仪进行分离纯化。
[0266]
色谱仪:岛津lc-20液相色谱仪(lc-20ad)。
[0267]
色谱柱:ymc-pack ods-aq,10mm*250mm,孔径5μm,日本ymc公司。
[0268]
流动相由三氟乙酸、乙腈和水组成,三氟乙酸的浓度为0.01%(体积比),乙腈的浓度为 90%(体积比)。流动相流速为2.5ml/min。
[0269]
检测波长:210nm。
[0270]
收集峰值对应的保留时间tr为19.8min的洗脱峰的过柱后洗脱液,采用旋转蒸发仪浓缩,然后进行冷冻干燥,得到化合物24(2.6mg)。
[0271]
实施例3、化学合成制备化合物
[0272]
一、化合物3和化合物4的制备
[0273]
反应物和产物以及收率见表1。反应过程:将10mg反应物a溶解于2ml反应物b(反应物b是过量的)中,加入阳离子交换树脂(dow 50wx8,50-100目(h),40mg),氩气保护环境中80℃热回流3-6小时(通过薄层色谱监测反应完全),然后采用4μm孔径滤膜过滤以除去树脂,收集滤液并进行减压浓缩以除去剩余的反应物b,获得产物c。
[0274]
表1
[0275][0276]
二、化合物13和化合物14的制备
[0277]
反应物和产物以及收率见表2。反应过程:将10mg反应物a溶解于2ml反应物b(反应物b是过量的)中,加入阳离子交换树脂(dow 50wx8,50-100目(h),40mg),氩气保护环境中80℃热回流3-6小时(通过薄层色谱监测反应完全),然后采用4μm孔径滤膜过滤以除去树脂,收集滤液并进行减压浓缩以除去剩余的反应物b,获得产物c。
[0278]
表2
[0279][0280]
三、化合物6的制备
[0281]
反应物和产物以及收率见表3。反应过程:将10mg反应物a和100μl反应物b(反应物b是过量的)溶解于2ml溶媒中,氩气保护环境中冰浴搅拌10min,然后依次加入9.5mg 1-乙基-3(3-二甲基丙胺)碳二亚胺和5mg 4-二甲氨基吡啶,先冰浴搅拌10min再室温搅拌5h,薄层监测反应完全,加入乙酸乙酯和水并静置分层,然后收集乙酸乙酯相并减压浓缩除去溶剂,获得产物c。
[0282]
表3
[0283][0284][0285]
四、化合物7、化合物8和化合物9的制备
[0286]
反应物和产物以及收率见表4。反应过程:将8.5mg反应物a溶解于2ml溶媒中,加入 20μl反应物b和21.9mg碳酸铯,氩气保护缓冲中室温搅拌2h;薄层监测反应完全后,加入乙酸乙酯和水并静置分层,然后收集乙酸乙酯相并减压浓缩除去溶剂,纯化得到产物c。
[0287]
表4
[0288][0289]
五、化合物25的制备
[0290]
反应物和产物以及收率见表5。反应过程:将5mg反应物a溶解于2ml溶媒中,加入 20mg反应物b和13.8mg碳酸铯,氩气保护环境中室温搅拌2h;薄层监测反应完全后,加入乙酸乙酯和水并静置分层,然后收集乙酸乙酯相并减压浓缩除去溶剂,纯化得到产物c。
[0291]
表5
[0292][0293]
实施例4、化合物的鉴定
[0294]
实施例2和实施例3中制备的化合物1至化合物27的外观:化合物1、化合物2、化合物4、化合物6、化合物7、化合物8、化合物9、化合物10、化合物13、化合物14和化合物25均为无色油状物;其余化合物均为白色无定型粉末。实施例2和实施例3中制备的化合物1至化合物27的溶解性:均易溶于甲醇且均易溶于乙腈。
[0295]
质谱:hresims测试采用thermo scientific ltq orbitrap xl质谱系统,甲醇为溶剂。
[0296]
对化合物的核磁共振谱进行研究并对1h-nmr和
13
c-nmr信号进行归属。
[0297]
各个化合物的hresi-ms谱、1h-nmr谱、
13
c-nmr谱见图1至图54。
[0298]
各个化合物的1h和
13
c nmr谱各峰归属见表6至表12。
[0299]
表6化合物1-5 1
h和
13
c nmr谱各峰归属(1h nmr 600mhz;
13
c nmr 150mhz;dmso-d6)
[0300]
[0301][0302]
表7化合物6-9 1
h和
13
c nmr谱各峰归属(1h nmr 600mhz;
13
c nmr 150mhz;dmso-d6)
[0303]
[0304][0305]
表8化合物10-14 1
h和
13
c nmr谱各峰归属(1h nmr 600mhz;
13
c nmr 150mhz;dmso-d6)
[0306][0307]
表9化合物15-17 1
h和
13
c nmr谱各峰归属(1h nmr 600mhz;
13
c nmr 150mhz;dmso-d6)
[0308][0309]
表10化合物18-21 1
h和
13
c nmr谱各峰归属(1h nmr 600mhz;
13
c nmr 150mhz;dmso-d6)
[0310]
[0311][0312]
表11化合物22-25 1
h和
13
c nmr谱各峰归属(1h nmr 600mhz;
13
c nmr 150mhz;cdcl3)
[0313]
[0314][0315]
表12化合物26-27 1
h和
13
c nmr谱各峰归属(1h nmr 600mhz;
13
c nmr 150mhz;cdcl3)
[0316]
[0317][0318]
根据鉴定结果,化合物1-27的结构式如下:
[0319][0320]
实施例5、化合物1-27的抗病毒活性
[0321]
一、对寨卡病毒的抗病毒活性
[0322]
供试化合物分别为实施例2和实施例3中制备的化合物1-17。利巴韦林(ribavirin)作为阳性对照。细胞培养条件均为:5%co2、37℃培养箱中。寨卡病毒(plcal_zv zika virusstrain)记载于如下文献:gao q,wang z,liu z,et al.identification oftheaflavin-3,3
’‑
digallate as a novel zika virus protease inhibitor[j].front. pharmacol.,2020,11:514313.doi:10.3389/fphar.2020.514313.ecollection。
[0323]
1、计算ic
50
值。
[0324]
(1)采用含10%fbs的dmem培养液悬浮vero细胞,得到8
×
104个细胞/ml的细胞悬液。
[0325]
(2)取96孔培养板,接种步骤(1)得到的细胞悬液,100μl/孔,培养24小时。
[0326]
(3)完成步骤(2)后,吸弃上清,每孔加入含供试化合物和10%fbs的dmem培养液,然后以moi=0.01的感染剂量接种寨卡病毒,然后培养96小时。设置不同的供试化合物浓
a virus[j].actapharmaceutica sinica b,2014,4(4):301-306。
[0338]
1、计算ic
50
值。
[0339]
(1)用含10%fbs的dmem培养液悬浮293t-gluc细胞,得到2.0
×
105个细胞/ml的细胞悬液。
[0340]
(2)取96孔培养板,接种步骤(1)得到的细胞悬液,100μl/孔,培养24小时。
[0341]
(3)完成步骤(2)后,每孔加入1μl供试化合物溶液,培养2小时。
[0342]
设置不同的供试化合物浓度(在体系中的浓度),具体浓度为:5、10、20、50、100或 200μm。
[0343]
供试化合物溶液是将供试化合物储存液用含10%fbs的dmem培养液稀释得到的。
[0344]
供试化合物储存液是将供试化合物溶于dmso得到的,化合物浓度为20mm。
[0345]
设置用等体积dmso代替供试化合物溶液的阴性对照。
[0346]
(4)完成步骤(3)后,以moi=0.3的感染剂量感染流感病毒a/wsn/33株,培养24小时。
[0347]
(5)完成步骤(4)后,每孔取样10μl上清,于多功能酶标仪(berthold centro lb960)测定荧光素酶活性,计算ic
50
值(将病毒抑制50%所需的浓度),结果见表14。
[0348]
2、计算cc
50
值。
[0349]
(1)用含10%fbs的dmem培养液悬浮293t-gluc细胞,得到2.0
×
105个细胞/ml的细胞悬液。
[0350]
(2)取96孔培养板,接种步骤(1)得到的细胞悬液,100μl/孔,培养24小时。
[0351]
(3)完成步骤(2)后,每孔加入1μl供试化合物溶液,培养48小时。
[0352]
设置不同的供试化合物浓度(在体系中的浓度),具体浓度为:5、10、20、50、100或 200μm。
[0353]
供试化合物溶液是将供试化合物储存液用含10%fbs的dmem培养液稀释得到的。
[0354]
供试化合物储存液是将供试化合物溶于dmso得到的,化合物浓度为20mm。
[0355]
设置用等体积dmso代替供试化合物溶液的阴性对照。
[0356]
(4)完成步骤(3)后,每孔加入10μl cck-8,37℃继续孵育1-2h后,使用多功能酶标仪(berthold centro lb 960)检测各孔在450nm波长处的光吸收值,用以计算半数细胞毒性浓度cc
50
(致使50%细胞死亡的药物浓度),结果见表14。
[0357]
表14化合物18-27抗甲型流感病毒活性评价
[0358] ic
50
(μm)cc
50
(μm)化合物185.53
±
1.10>100化合物1913.06
±
0.09>100化合物2010.78
±
1.03>100化合物212.90
±
0.30>100化合物2212.45
±
1.40>100化合物2411.25
±
0.64>100化合物2412.46
±
0.15>100化合物2512.94
±
0.86>100化合物2611.26
±
0.00>100
化合物2715.02
±
0.05>100ribavirin21.72
±
0.38>100
[0359]
结果显示,各个化合物对人甲型流感病毒复制均有抑制活性,其ic
50
值在2.9-15.02μm 之间,且在100μm浓度下无明显细胞毒性,对流感病毒的抑制作用高于阳性药利巴韦林(ribavirin)。说明本发明的化合物对流感病毒具有较强的抑制活性,并且毒性较小。
[0360]
以上对本发明进行了详述。对于本领域技术人员来说,在不脱离本发明的宗旨和范围,以及无需进行不必要的实验情况下,可在等同参数、浓度和条件下,在较宽范围内实施本发明。虽然本发明给出了特殊的实施例,应该理解为,可以对本发明作进一步的改进。总之,按本发明的原理,本技术欲包括任何变更、用途或对本发明的改进,包括脱离了本技术中已公开范围,而用本领域已知的常规技术进行的改变。按以下附带的权利要求的范围,可以进行一些基本特征的应用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1