一种分级结构微球药物载体的制备方法与流程
:
1.本发明属于生物医用材料技术领域,具体涉及一种基于微流控电喷技术的分级结构微球药物载体的制备方法。
背景技术:
2.癌症现在已成为世界上导致死亡的主要因素。化疗作为最有效的治疗手段之一,通过直接杀死癌细胞在肿瘤学领域取得了巨大的成功。近年来,肿瘤免疫治疗是一种突破性的治疗策略,通过刺激宿主免疫系统来实现肿瘤的消退,已经彻底改变了临床肿瘤的治疗。然而,单纯化疗或免疫治疗在癌症治疗中疗效不佳。一方面,化疗药物选择性差,循环时间短,生物利用度低,极大地限制了化疗的有效性。另一方面,免疫抑制微环境、肿瘤免疫原性低、肿瘤内细胞毒性t淋巴细胞浸润不足导致目前免疫治疗的临床免疫应答率低。此外,传统的全身化疗作为一种常用的应用模式,会对正常组织造成严重的副作用和不可避免的全身毒性。同时,全身给药后免疫治疗引起非特异性免疫反应时,常发生危险的细胞因子风暴等副作用。因此,迫切需要开发一种合适的配方,以实现高效的化疗免疫治疗,同时减少全身毒副作用。
3.纳米技术的快速发展可以实现多种药物的共同递送,以提高药物联合治疗的效果。然而,纳米载体在临床试验中仍远未达到令人满意的效果,因为肿瘤蓄积不足,系统给药后可能出现药物过量导致毒副作用严重。基于微流控微载体的系统具有良好的可注射性、在肿瘤部位有足够的药物积累以及降低对健康组织的毒性等诸多优点,改善了给药方式和肿瘤治疗的效果。但仍难以解决不同药物或载体材料之间溶解度不同的矛盾,导致载药过程十分困难。
4.因此,在本发明中,我们以单药聚合物或者双药聚合物为原料,通过微流控电喷技术制备了一种分级结构微球药物载体,可用于局部药物递送,实现高效联合治疗效果并降低药物毒副作用。
技术实现要素:
5.本发明的目的是针对现有技术的不足,提供一种分级结构微球药物载体的制备方法,解决了传统系统给药方式选择性差、循环时间短、生物利用度低,严重的副作用和非特异性免疫反应等问题,具有制备方法简单、材料生物相容性好、药物结构明确、成本低廉、药物负载率高的优势,同时还可避免突释并隐匿药物毒副作用等优点,可应用于肿瘤局部高效药物递送及联合治疗领域。
6.为实现上述技术目的,本发明采用以下技术方案:
7.一种分级结构微球药物载体的制备方法,包括以下步骤:
8.s1、将四价奥沙利铂前药oxapt(iv)作为聚合单体合成主链含铂高分子,进一步在主链含铂高分子侧链上键合聚乙二醇/小分子ido1抑制剂,得到担载单药/双药的聚合物,并将单药/双药聚合物通过自组装制备得到载药聚合物纳米粒;
9.s2、将s1得到的载药聚合物纳米粒与海藻酸钠进行共混并搅拌均匀,随后通过微流控电喷至氯化钙溶液中,得到担载单药/双药聚合物纳米粒的分级结构微球。
10.进一步的,所述s1中,主链含铂高分子合成方法为:将四价奥沙利铂前药oxapt(iv)与环丁烷四甲酸二酐(cbda)混合,加入n,n-二甲基甲酰胺(dmf),混合搅拌反应,得到主链含铂高分子(polyoxapt)的反应溶液。
11.进一步的,四价奥沙利铂前药oxapt(iv)与环丁烷四甲酸二酐(cbda)摩尔比为1:1;四价奥沙利铂前药oxapt(iv)在n,n-二甲基甲酰胺(dmf)中的浓度为17.24mg/ml;环丁烷四甲酸二酐(cbda)在n,n-二甲基甲酰胺(dmf)中的浓度为8.04mg/ml;反应温度为80℃,反应时间为3天。
12.进一步的,所述s1中,担载单药聚合物的制备方法具体为:将二环己基碳二亚胺dcc和4-二甲氨基吡啶dmap加入到主链含铂高分子(polyoxapt)的反应溶液中,在冰水浴中搅拌,加入mpeg
2k-oh并继续搅拌,过滤,将溶液沉降于冰乙醚中,随后将沉降物用dmf溶解后置于透析袋中进行透析,透析后冻干,即得到单药聚合物(polyoxapt-peg,sdp);其中,四价奥沙利铂前药oxapt(iv)、二环己基碳二亚胺、4-二甲氨基吡啶、mpeg
2k-oh的摩尔比为1:2:2:1。
13.进一步的,冰水浴中搅拌时间为1h,加入mpeg
2k-oh后搅拌24h,透析时间为72h,透析袋截留分子量为3500。
14.进一步的,所述s1中,担载双药聚合物的制备具体为:将二环己基碳二亚胺dcc和4-二甲氨基吡啶dmap加入到主链含铂高分子(polyoxapt)的反应溶液中,在冰水浴中搅拌,接着加入ido1抑制剂在室温下继续搅拌反应,随后加入mpeg
2k-oh并搅拌,过滤,将溶液沉降于冰乙醚中,将沉降物用dmf溶解后置于透析袋中进行透析,透析后冻干,即得到双药聚合物(polyoxapt-nlg919/peg,ddp);其中,四价奥沙利铂前药oxapt(iv)、二环己基碳二亚胺、4-二甲氨基吡啶、ido1抑制剂、mpeg
2k-oh的摩尔比为1:2:2:1:1。
15.进一步的,冰水浴中搅拌时间为1h,加入ido1抑制剂后搅拌12h,加入mpeg
2k-oh后搅拌24h,透析时间为72h,透析袋截留分子量为3500。
16.进一步的,所述ido1抑制剂为nlg919。
17.进一步的,所述s1中,自组装制备载药聚合物纳米粒具体为:将双药聚合物/单药聚合物溶于n,n-二甲基甲酰胺(dmf)中,边搅拌边逐滴加入水,使双药聚合物/单药聚合物自组装成纳米粒,随后通过透析除去溶液中多余的小分子溶剂,冻干后得到双药聚合物纳米粒(ddp nps)或者单药聚合物纳米粒(sdp nps)。
18.进一步的,双药聚合物/单药聚合物的n,n-二甲基甲酰胺(dmf)溶液中,双药聚合物/单药聚合物的浓度为5mg/ml,所滴加的水与n,n-二甲基甲酰胺(dmf)的体积比为15:2;透析时间为2天,透析截留分子量为3500。
19.进一步的,所述s2中,分级结构微球的制备方法具体为:
20.s2.1、将双药聚合物纳米粒(ddp nps)/单药聚合物纳米粒(sdp nps)和海藻酸钠(naalg)溶于水中,搅拌均匀,得到预凝胶溶液;
21.s2.2、将预凝胶溶液注入微流控装置中,通过注射泵及高压电喷将溶液泵出至cacl2溶液中,得到分级结构微球药物载体。
22.进一步的,注射泵流速为2ml h-1
,高压电场为4kv。
23.进一步的,预凝胶溶液中,所述双药聚合物纳米粒(ddp nps)/单药聚合物纳米粒(sdp nps)的质量体积浓度为2%,海藻酸钠(naalg)的质量体积浓度为2%;cacl2溶液中,cacl2的的质量体积浓度为2%。
24.进一步的,所制备的分级结构微球药物载体为担载单药聚合物纳米粒的微球或担载双药聚合物纳米粒的微球;所述担载单药聚合物纳米粒的微球直径为145
±
14μm,微球内部含有大量单药聚合物纳米粒;所述担载双药聚合物纳米粒的微球直径为144
±
13μm,微球内部含有大量双药聚合物纳米粒。
25.本发明的有益效果:
26.(1)采用本发明方法所制备的双药聚合物纳米粒(ddp nps)/单药聚合物纳米粒(sdp nps),在主链含铂高分子侧链键合上小分子ido1抑制剂以及聚乙二醇,药物结构明确,药物含量高,相较于普通的药物包裹,具有更好的稳定性;
27.(2)本发明以微流控电喷得到的分级结构微球药物载体具有制备简单、成本低廉、材料生物相容性好、操作方便,不需要很高的技术要求,且易对微球的大小进行控制;
28.(3)本发明所制备的分级结构微球药物载体药物,负载率高,缓释效果好,可避免突释并隐匿药物毒副作用;
29.(4)本发明制备的分级结构微球药物载体为可注射药物载体,可用于肿瘤局部药物递送,实现了长时间药物病灶部位蓄积和药物缓释,可自主给药,实用性强,可应用于肿瘤局部高效联合治疗领域。
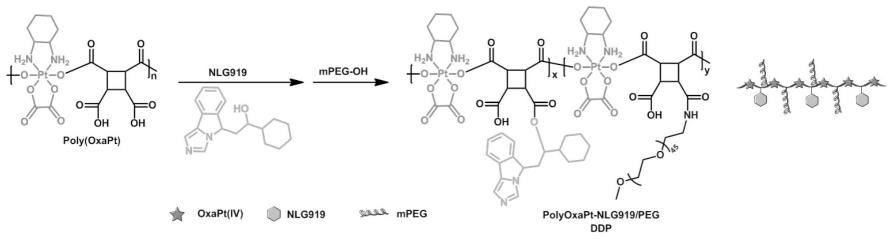
附图说明:
30.图1为本发明实施例双药聚合物(ddp)制备示意图;
31.图2为本发明实施例双药聚合物纳米粒(ddp nps)制备示意图;
32.图3为本发明实施例担载双药聚合物纳米粒的分级结构微球药物载体(m
ddp
)制备示意图;
33.图4为本发明实施例所制备的不担载纳米粒的微球(m
blank
)及担载单药聚合物纳米粒的分级结构微球药物载体(m
sdp
)示意图;
34.图5为本发明实施例所制备的单药聚合物(sdp)和双药聚合物(ddp)的核磁表征;
35.图6为本发明实施例所制备的单药聚合物纳米粒(sdp nps)和双药聚合物纳米粒(ddp nps)的透射电镜形貌图;
36.图7为本发明实施例所制备的m
blank
,m
sdp
及m
ddp
的光学形貌、扫描电镜形貌、内部结构及粒径分析图;
37.图8为本发明实施例所制备的m
ddp
释放的ddp nps(a)及ddp nps被降解(b)的透射电镜图;
38.图9为本发明实施例所制备的m
ddp
在不使用透析袋(a)和使用透析袋(b)和的情况下药物的释放情况。
具体实施方式:
39.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是
本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。下述实施例中所使用的实验方法,如无特殊说明,均为常规方法,所用的试剂、方法和设备,如无特殊说明,均为本技术领域常规试剂、方法和设备。
40.实施例1——担载双药聚合物纳米粒的分级结构微球药物载体的制备。
41.(一)、主链含铂高分子的制备:
42.合成z轴配体为双羟基的四价奥沙利铂前药oxapt(iv),并将其与1,2,3,4-环丁烷四甲酸二酐(cbda)进行开环聚合反应制备得到主链含oxapt(iv)的聚前药高分子(polyoxapt)。具体地,将四价奥沙利铂前药oxapt(iv)(86.2mg,0.2mmol)和环丁烷四甲酸二酐(cbda,40.2mg,0.205mmol)加入到干燥的聚合瓶中,随后加入5ml干燥的dmf,在80℃下搅拌反应3天,冷却至室温,得到主链含铂高分子(polyoxapt)的反应溶液。
43.(二)、担载双药聚合物(ddp)的制备:
44.将ido1抑制剂(nlg919)以及聚乙二醇分别键合至polyoxapt侧链,得到主链含铂、侧链含nlg919的两亲性高分子前药(polyoxapt-nlg919/peg,ddp)。具体的,将dcc(82.5mg,0.4mmol)和dmap(48.9mg,0.4mmol)加入到主链含铂高分子(polyoxapt)的反应溶液中,在冰水浴中搅拌1h,然后将nlg919(56.5mg,0.2mmol)在室温下继续搅拌反应12h。随后,在溶液中加入mpeg
2k-oh(100mg,0.2mmol)搅拌反应24小时,过滤除去二环己基脲(dcu)后,将溶液沉降于冰乙醚中。将沉降物用2ml dmf溶解后置于透析袋(mwco=3500)中,在去离子水中透析72h后冻干,即得到双药聚合物(polyoxapt-nlg919/peg,ddp)(如图1)。通过核磁共振波谱对ddp进行表征,结果如图5所示。
45.(三)、双药聚合物纳米粒(ddp nps)的制备:
46.首先将ddp(20mg)溶于4ml dmf中,在搅拌下缓慢滴加30ml h2o,使双药聚合物自组装成纳米粒;随后将纳米粒溶液加入透析袋(mwco=3500)中在去离子水中透析2天后冻干,即得到双药聚合物纳米粒ddp nps(如图2)。采用透射电镜对制备的ddp nps进行表征,结果如图6所示。
47.(四)、担载双药聚合物纳米粒的分级结构微球药物载体(m
ddp
)的制备:
48.将ddp nps(2%,w/v)和海藻酸钠(naalg,2%,w/v)溶于水中,搅拌混匀得到预凝胶溶液;将该预凝胶溶液吸入注射器后通过注射泵泵入微流体装置,设置注射泵流速和电压分别为2ml h-1
和4kv,将泵出的溶液收集在cacl2(2%,w/v)中得到分级结构微球药物载体(m
ddp
)(如图3),其直径为144
±
13μm。采用光学显微镜,扫描电镜对m
ddp
的形貌进行表征,结果如图7c、f所示。
49.实施例2——担载单药聚合物纳米粒的分级结构微球药物载体的制备。
50.(一)、主链含铂高分子的制备:
51.将四价奥沙利铂前药oxapt(iv)(86.2mg,0.2mmol)和环丁烷四甲酸二酐(cbda,40.2mg,0.205mmol)加入到干燥的聚合瓶中,随后加入5ml干燥的dmf,在80℃下搅拌反应3天,冷却至室温,得到主链含铂高分子(polyoxapt)的反应溶液。
52.(二)、担载单药聚合物(sdp)的制备:
53.将dcc(82.5mg,0.4mmol)和dmap(48.9mg,0.4mmol)加入到主链含铂高分子(polyoxapt)的反应溶液中,在冰水浴中搅拌1h,然后在溶液中加入mpeg
2k-oh(100mg,
0.2mmol)搅拌反应24小时,过滤除去dcu后,将溶液沉降于冰乙醚中。将沉降物用2ml dmf溶解后置于透析袋(mwco=3500)中,在去离子水中透析72h后冻干,即得到单药聚合物(polyoxapt-peg,sdp)。通过核磁共振波谱对sdp进行表征,结果如图5所示。
54.(三)、单药聚合物纳米粒(sdp nps)的制备:
55.通过纳米共沉淀的方法制备得到单药聚合物纳米粒。具体操作如下:首先将sdp(20mg)溶于4ml dmf中,在搅拌下缓慢滴加30ml h2o,使单药聚合物自组装成纳米粒;随后将纳米粒溶液加入透析袋(mwco=3500)中在去离子水中透析2天后冻干,即得到单药聚合物纳米粒sdp nps。采用透射电镜对制备的sdp nps进行表征,结果如图6所示。
56.(四)、担载单药聚合物纳米粒的分级结构微球药物载体(m
sdp
)的制备:
57.将sdp nps(2%,w/v)和海藻酸钠(naalg,2%,w/v)溶于水中,搅拌混匀得到预凝胶溶液。将该预凝胶溶液吸入注射器后通过注射泵泵入微流体装置,设置注射泵流速和电压分别为2ml h-1
和4kv,将泵出的溶液收集在cacl2(2%,w/v)中得到分级结构微球药物载体(m
sdp
)(如图4),其直径为145
±
14μm。采用光学显微镜,扫描电镜对m
sdp
的形貌进行表征,结果如图7b、e所示。
58.效果试验
59.将实施例1所制备的载药微球(m
ddp
)进行体外药物释放效果研究。
60.为了研究纳米粒从微球中的释放及纳米粒的还原响应性,我们通过在使用透析袋和不使用透析袋的情况下,分别研究了m
ddp
中纳米粒的释放以及进一步pt和nlg919的释放行为。
61.(1)通过在未使用透析袋的情况下说明纳米粒从微球中的释放,m
ddp
直接孵育在pbs中,随后置于37℃振荡培养箱中。在预先确定的时间点后,收集1ml透析液,并加入1ml pbs保证体系体积不变。收集的透析液用icp-ms和紫外-可见分光光度计分别测定pt和nlg919的释放量。从图8a投射电镜图可以看出,m
ddp
释放出的纳米粒ddp nps可以被检测到。且由于释放的是纳米粒和小分子pt和nlg919,从释放结果可以看到(如图9a所示),有将近68.26%的pt和64.04%的nlg919在前10h内可以被检测到,并且在96h内93.87%的pt和95.53%的nlg919可以检测到。
62.(2)通过在使用透析袋的情况下说明从微球中的释放的纳米粒的还原响应性,首先将m
ddp
加入透析袋(mwco=1000)中,然后在试管中分别浸泡于19ml不同的释放介质:pbs(不含gsh)、pbs(含有10mm gsh,gsh具有还原性,能将纳米粒中的高分子还原降解),置于37℃振荡培养箱中。在预先确定的时间点后,收集1ml透析液,并加入1ml pbs保证体系体积不变。收集的透析液用icp-ms和紫外-可见分光光度计分别测定pt和nlg919的释放量。从图8b投射电镜图可以看出,纳米粒ddp nps被还原降解的小分子pt和nlg919可以被检测到。从释放结果可以看到(如图9b所示),在pbs释放介质中,pt和nlg919从微球中缓慢释放,在96h后分别仅检出18.16%和22.06%的pt和nlg919。相比之下,在含有gsh的释放介质中,由于gsh对纳米粒的还原降解作用,药物释放速度更快,释放的pt和nlg919在96h内可达到73.31%和75.87%,说明ddp nps的还原响应性。
63.本发明方法所制备的双药聚合物纳米粒(ddp nps)或单药聚合物纳米粒(sdp nps),药物结构明确,药物含量高,相较于普通的药物包裹,缓释效果好,可避免突释并隐匿药物毒副作用,具有更好的稳定性。本发明所制备的分级结构微球药物载体为可注射药物
载体,可用于肿瘤局部药物递送,实现了长时间药物病灶部位蓄积和药物缓释,可应用于肿瘤局部高效联合治疗领域。
64.以上仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1