一种天麻微囊及其制备方法与应用
1.本发明属于中药提取制备技术领域,具体涉及一种天麻微囊及其制备方法与应用。
背景技术:
2.天麻是与真菌共生的草本植物,对生长环境的要求高,喜潮湿阴凉的环境,需要依托良好的杂木混交林和充足的水分,天麻多生长于林地较多的高海拔山区(800~2500米),最高温度不超过30℃,空气湿度不低于50%,年降水量在1000mm左右,地形宜选择在背阴坡地。我国的西南地区为天麻的主产区,包括贵州、云南、四川、陕西、湖北等地,天麻中富含百余种化学成分,其中活性成分主要有酚类及其苷类、有机酸类、甾醇类、多糖类等。
3.天麻的提取方法主要有渗漉法、回流提取法、酶解提取法、超声提取法和微波提取法,传统提取方法对天麻的提取分离存在提取时间长、温度高、热敏成分易降解等问题,不同提取分离技术的特点见表1。
4.表1天麻的传统提取分离技术优缺点
[0005][0006]
低温高压破碎提取技术相比于传统提取方法,提取时间较短,提取效率高,而且对热敏性成分有良好的提取效果。其原理是在低温条件下,施加流体动态高压,形成瞬间高压,再瞬间降压,产生强烈的剪切、湍流及冲击等作用,使物料细胞瞬间膨胀并破碎,加大有效成分与溶剂接触面积,加速溶剂渗透和成分释放,在破碎细胞的同时达到高效提取的目的。高压破碎提取技术可将破壁与提取相结合,整个提取过程均在低温及连续的条件下进行,相比于传统机械破碎方法,可避免热敏性活性成分遇热降解或失活的等问题,且工艺简单,高效节能,可降低生产成本。
[0007]
目前的天麻制剂多以天麻中的单体成分天麻素制剂为主,多为传统制剂,包括片剂、注射剂、滴丸和软胶囊等。文献《天麻素缓释片的制备及其体外释药特性研究》采用渗透泵技术,以醋酸纤维素作为包衣材料,制备了天麻素缓释片;文献《聚山梨酯80修饰的天麻素脂质体制备研究》采用薄膜-超声法制备得到了能够跨过血脑屏障的聚山梨酯80修饰的
天麻素脂质体注射液;《天麻素纳米脂质体和纳米微球的制备研究》采用溶剂挥发-复乳法制备得到了天麻素微球。然而中药药效多为多种成分综合作用的结果,目前关于天麻提取物多成分制剂的研究较少,能避免热敏性成分遭到破环和损失的微胶囊剂的研究极少。文献《天麻提取物微胶囊的制备》以海藻酸钠为壁材,采用滴制法制备得到了粒径为200μm、包埋率为48%左右的天麻提取物海藻酸钙微囊,其存在制备工艺耗时长且天麻素的包埋率较低问题。
技术实现要素:
[0008]
为解决相关问题,本发明的首要目的在于提供一种天麻微囊的制备方法。
[0009]
本发明的另一目的在于提供通过上述制备方法得到的天麻微囊。
[0010]
本发明的再一目的在于提供上述天麻微囊的应用。
[0011]
为了实现上述发明目的,本发明采用以下技术方案:
[0012]
一种天麻微囊的制备方法,包括如下步骤:
[0013]
步骤(1):低温高压破碎提取:将天麻粉加入提取溶剂中,用高速匀浆机均匀分散,注入高压破碎提取装置内进行破碎提取,提取物离心,取滤液;其中,提取溶剂为体积分数为30%~70%的乙醇,液固比10:1~40:1,破碎提取的温度3~5℃、压力50~200mpa,次数为1~4次;
[0014]
步骤(2):冷冻干燥:将步骤(1)所得滤液进行冷冻干燥,干燥至恒重,得到天麻提取物冻干粉;
[0015]
步骤(3):高压均质-喷雾干燥法制备微囊:将壁材和乳化剂加入水中,搅拌均匀后加入作为芯材的步骤(2)得到的天麻提取物冻干粉;先用高速分散机进行高速搅拌,再用高压均质机均质,制备得到天麻均质液,天麻均质液进行喷雾干燥,得天麻微囊。
[0016]
进一步地,步骤(1)中所述的高速匀浆机的转速为10000r/min,均匀分散的时间为2min。
[0017]
进一步地,步骤(1)中所述的离心的转速为4000r,时间为5min。
[0018]
进一步地,步骤(1)中提取溶剂为体积分数为50%的乙醇,液固比30:1,破碎提取的温度4℃、压力100mpa。
[0019]
进一步地,步骤(3)中所述的天麻提取物冻干粉中天麻素质量百分含量0.281%、对羟基苯甲醇质量百分含量1.509%、巴利森苷e质量百分含量0.964%、巴利森苷b质量百分含量0.717%、巴利森苷c质量百分含量0.123%、巴利森苷a质量百分含量2.422%。
[0020]
进一步地,步骤(3)中所述的壁材为大豆分离蛋白(spi)和麦芽糊精(md)组成的复合壁材,大豆分离蛋白和麦芽糊精的配比为质量比0.5:1~2.5:1,优选为质量比1:1。
[0021]
进一步地,步骤(3)中所述的乳化剂为单甘脂。
[0022]
进一步地,步骤(3)中所述的壁材的质量浓度为2%~10%、乳化剂的质量浓度为0.6%~0.8%、壁材和芯材的质量比为3:1~7:1;优选的,所述的壁材的质量浓度为8%、乳化剂的质量浓度为0.67%、壁材和芯材的质量比为7:1。
[0023]
进一步地,步骤(3)中所述的喷雾干燥的进料速度为最大进料速度的10%~30%,最大进料速度为1.5ml/min。
[0024]
进一步地,步骤(3)中所述的高速分散机的转速为10000r/min,高速搅拌2min。
[0025]
进一步地,步骤(3)中所述的高压均质机的压力为50mpa。
[0026]
一种天麻微囊,通过上述制备方法得到。
[0027]
进一步地,所述的天麻微囊的粒径分布为1~10μm,包封率达到87.48%,收率达到75.96%,载药量达到15.46%。
[0028]
上述天麻微囊在制备肠道释放药物中的应用。
[0029]
本发明相对于现有技术具有如下的优点及效果:
[0030]
本发明公开了一种天麻微囊的制备方法,采用低温高压破碎提取技术结合冷冻干燥技术制备天麻提取物,天麻冻干粉再采用高压均质-喷雾干燥技术制备成微囊。与回流提取-减压干燥技术相比,本方法所得的天麻冻干粉的总收率、目标成的成分的提取率均高于传统方法,具有更高的抗氧化能力;采用高压均质-喷雾干燥技术制备的微囊包封率高、收率高、载药量大,且载药后提高了天麻中热敏性成分的热稳定性,且具有一定的缓释作用,有利于提高天麻中活性成分的生物利用度,为天麻的有效利用及制剂开发提供依据。
附图说明
[0031]
图1为乙醇浓度对天麻中6种指标成分提取率的影响研究结果图;
[0032]
图2为液固比对天麻中6种指标成分提取率的影响研究结果图;
[0033]
图3为提取压力对天麻中6种指标成分提取率的影响研究结果图;
[0034]
图4为提取次数对天麻中6种指标成分提取率的影响研究结果图;
[0035]
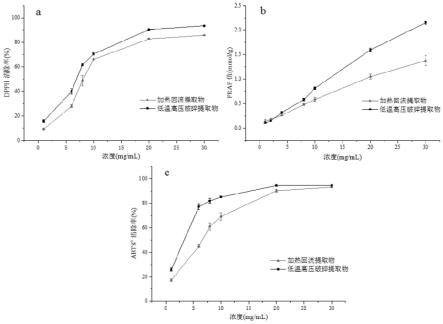
图5为加热回流提取物和低温高压破碎提取物的抗氧化能力研究结果图;其中,a为dpph自由基清除试验结果,b为frap试验结果,c为abts自由基清除试验结果;
[0036]
图6为不同样品的扫描电镜图;其中,a为麦芽糊精,b为大豆分离蛋白,c为天麻冻干粉,d为物理混合物,e为天麻提取物微囊;
[0037]
图7为微囊粒径分布图;
[0038]
图8为傅里叶红外光谱图;
[0039]
图9为x射线衍射光谱图;其中,a为天麻提取物冻干粉,b为天麻提取物微囊,c为大豆分离蛋白,d为麦芽糊精;
[0040]
图10为热重分析(tg)图;其中,a为天麻取物冻干粉,b为麦芽糊精,c为大豆分离蛋白,d为天麻提取物微囊;
[0041]
图11为差示扫描量热(dsc)图;
[0042]
图12为不同温度下天麻提取物微囊和物理混合物的相对含量曲线图;其中,a为40℃,b为60℃,c为80℃;
[0043]
图13为天麻低温提取物冻干粉及天麻低温提取物微囊的体外溶出曲线图。
具体实施方式
[0044]
为更好地理解本发明,下面通过以下实施例对本发明作进一步具体的阐述,但不可理解为对本发明的限定,对于本领域的技术人员根据上述发明内容所作的一些非本质的改进与调整,也视为落在本发明的保护范围内。
[0045]
本发明实施例中涉及的成分检测分析方法:
[0046]
(1)色谱条件
[0047]
色谱柱:phenomenex luna c18(250mm
×
4.6mm,5μm);流动相:乙腈(a)-0.1%磷酸水(b),梯度洗脱(0~5min,3.0%a;5~15min,3.0%~5.0%a;15~22min,5.0%a;22~25min,5.0%~10.1%a;25~35min,10.1%~10.2%a;35~45min,10.2%~14.0%a;45~52min,14.0%a;52~55min,14.0%~16.5%a;55~63min,16.5%~17.5%a;63~65min,17.5%~20.0%a;65~70min,20.0%a);流速:1.0ml/min;检测波长:220nm;柱温:35℃;进样量:4μl。
[0048]
(2)对照品溶液的制备
[0049]
精密称取天麻素、对羟基苯甲醇、巴利森苷e、巴利森苷b、巴利森苷c和巴利森苷a对照品适量,加30%色谱级乙腈溶液,制成浓度分别为0.083 0、0.069 4、0.084 5、0.076 0、0.027 3、0.207 0mg/ml的混合对照品溶液,过0.45μm的微孔滤膜,备用。
[0050]
(3)供试品溶液的制备
[0051]
称取天麻冻干粉或微囊适量,置于50ml具塞锥形瓶中,加入适量甲醇,超声处理40min(40khz,250w),冷却后,再称定重量,用甲醇补足失重,摇匀,即得。
[0052]
(4)线性关系及范围
[0053]
配制不同浓度的混合对照品溶液,采用上述色谱条件测定峰面积,以各对照品的质量为横坐标,相应的峰面积值为纵坐标,绘制标准曲线,各标准曲线的回归方程、相关系数、线性范围、定量限、检出限结果见表2。结果表明,6个指标成分在相应范围内线性关系良好。
[0054]
表2 6个指标成分的线性方程、相关系数和线性范围
[0055][0056]
实施例1:低温高压破碎提取
[0057]
1、乙醇浓度的影响
[0058]
溶剂性质在萃取过程中起着至关重要的作用,它受溶剂极性、溶液粘度和表面张力的影响,单一溶剂(水)可能不能有效地提取所需的所有成分。因此,有机溶剂被用来提高萃取效率。选用绿色溶剂乙醇水溶液作为提取溶剂。结果如图1所示。随着乙醇浓度从10%增加到50%,目标化合物的提取率增加。
[0059]
2、液固比的影响
[0060]
结果如图2所示。当液固比从10:1增加到30:1时,目标化合物的提取率总和有所提高。在此之后,提取率随着液固比的升高而降低。各单一成分的变化趋势基本相同。
[0061]
3、提取压力的影响
[0062]
结果如图3所示。随着提取压力从50mpa升高到200mpa,目标化合物的提取率总和
先增高后降低,提取率总和差异不大。天麻素、对羟基苯甲醇、巴利森苷e、巴利森苷c和巴利森苷a的提取率变化趋势基本相同。
[0063]
4、提取次数的影响
[0064]
结果如图4所示。随着低温高压破碎提取次数的增加,天麻中目标化合物的提取率总和逐渐降低,可能是由于提取次数增加,提取液有损失,各单一成分的变化趋势基本相同。
[0065]
5、低温高压破碎提取响应面试验
[0066]
由以上单因素结果可知,乙醇浓度、料液比、提取压力对于天麻目标成分的提取影响较大,对此三个因素采取box-behnken(bbd)设计方法,以六种成分收率作为响应值进行优化。
[0067]
采用三水平三因素bbd设计,确定不同自变量对天麻靶成分提取的影响及各自变量的综合效应。如表3所示,所选择的自变量为乙醇浓度(x1);液固比(x2);提取压力(x3)和这些变量的三阶乘水平分别为-1、0和+1,分别为低、中、高水平。因变量为目标化合物天麻素(y1)、对羟基苯甲醇(y2)、巴利森苷e(y3)、巴利森苷b(y4)、巴利森苷c(y5)和巴利森苷a(y6)的提取率。
[0068]
表3响应面分析因素和水平表
[0069][0070]
根据上述bbd试验,得到了表4所示的最佳操作参数。为方便实际操作,选择乙醇浓度为50%、液固比为30:1、提取压力为100mpa的条件对四种目标化合物进行低温高压破碎提取。在接下来的实验中采用了最优条件。
[0071]
表4通过bbd试验确定的目标化合物的最佳提取条件和提取率
[0072][0073]
对上述最佳工艺条件进行验证,结果见表5。6种指标成分的提取率之和为1.599%,其中天麻素、对羟基苯甲醇、巴利森苷e、巴利森苷b、巴利森苷c、巴利森苷a的提取率分别为0.068%、0.497%、0.249%、0.161%、0.036%和0.588%,与理论优化值无显著差异,说明所创建的响应面模型拟合度良好,适用于天麻中6种有效成分的提取工艺的优化。
[0074]
表5验证试验结果
[0075][0076]
对低温高压破碎提取滤液在-80℃的温度下进行冷冻干燥,干燥至恒重,得到天麻提取物冻干粉。
[0077]
对比例1:低温高压破碎提取与传统方法比较
[0078]
1、提取率对比
[0079]
低温高压破碎提取法(方法1):按照本发明方法进行低温高压破碎提取(温度4℃、时间5min)、离心、滤液进行冷冻干燥,干燥至恒重,即得到天麻低温高压破碎提取物。
[0080]
加热回流提取法(方法2):参考中国药典中天麻含量测定法项下方法,精密称定天麻粉末2g,放置于锥形瓶中,加入稀乙醇溶液50ml,称重,加热回流3h(温度80℃、时间180min),放冷,再称重,用稀乙醇补足失重,过滤,取滤液备用。将回流提取液在90℃,0.08mpa条件下进行真空旋蒸干燥,干燥至恒重,即得到天麻传统回流提取物。
[0081]
表6不同提取方法的比较
[0082][0083]
如表6所示,低温高压破碎提取天麻素、对羟基苯甲醇、巴利森苷e、巴利森苷b、巴
利森苷c和巴利森苷a的提取率高于传统回流提取。巴利森苷类化合物是热不稳定化合物,在高温下会发生降解,传统回流的提取方法需要较长的提取时间和较高的提取温度,不适合热不稳定化合物的提取。此外,低温高压破碎的提取时间仅为10min左右,远低于传统回流提取的提取时间(3h),提取时间过长会产生较大的损耗从而使转移率降低。因此,与传统提取方法比较,本发明具有更大的发展潜力和应用价值。
[0084]
2、抗氧化能力对比
[0085]
(1)dpph自由基清除试验:通过对dpph自由基的稳定清除,评价其抗氧化活性。将0.1ml样品溶液与1.4ml乙醇混合后,加入dpph(0.04mg/ml,1ml)。在黑暗中反应60min,用紫外分光光度计在517nm处测定样品溶液的吸光度。dpph清除率的计算公式如下:
[0086][0087]
dpph清除率越大、ic
50
值越小表示抗氧化能力越强,低温高压破碎提取物的ic
50
值为7.455mg/ml,小于加热回流提取物的ic
50
值(7.792mg/ml),结合图5中a的结果可得,低温高压破碎提取物的dpph自由基清除能力强于加热回流提取物。
[0088]
(2)frap试验:取8mltptz稀释液和0.8ml的tptz溶液混匀,加入0.8ml检测缓冲液,即得到frap工作液,在37℃条件下孵育备用。称取27.9mg feso4·
7h2o,溶解并定容到1ml,此时浓度即为100mm。取适量100mm feso4溶液稀释至0.15、0.3、0.6、0.9、1.2和1.5mm。以摩尔浓度(mm,x)为横坐标,吸光度(y)为纵坐标,绘制feso4的标准曲线。结果表明,feso4在0.15~1.5mm范围内呈良好的线性关系,线性回归方程为:y=0.3321x+0.0489(r2=0.9993)
[0089]
将不同浓度(1、6、8、10、20、30mg/ml)的样品溶液与frap工作液混合,在37℃孵育5min。用酶标仪在593nm处测定吸光度。样品的最终总抗氧化能力以feso4等效浓度(frap值)表示。frap值越大,抗氧化能力越强,低温高压破碎提取物的frap值为0.082mmol/g大于加热回流提取物的frap值0.074mmol/g,从图5中b中也可得出,前者的总抗氧化能力更强。
[0090]
(3)abts自由基清除试验:取0.2ml abts溶液和0.2ml氧化剂溶液混匀,得到abts工作母液,在室温下避光保存12小时后使用。取0.1ml abts工作母液,用80%的乙醇溶液稀释至50ml得到abts工作液。将不同浓度(1、6、8、10、20、30mg/ml)的样品与abts工作液混合,37℃下孵育5min。用酶标仪在734nm波长下测定吸光度。abts清除率的计算公式如下:
[0091][0092]
abts清除率越大,抗氧化能力越强,从图5中c可知低温高压破碎提取物的抗氧化能力更强,其ic
50
值为1.984mg/ml,小于加热回流提取物的ic
50
值(7.437mg/ml),因此,低温高压破碎提取物的abts自由基清除能力更强。
[0093]
表7传统回流提取物和低温高压破碎提取物的自由基清除活性
[0094][0095]
如表7所示,与加热回流提取物相比,低温高压破碎提取物具有更高的抗氧化能力,包括清除dpph自由基能力、frap铁离子还原能力和abts总抗氧化能力。这可能是与天麻的低温高压破碎提取物中巴利森苷类化合物的含量更高有关。
[0096]
实施例2:天麻提取冻干粉微囊研究
[0097]
1、壁材浓度对微囊成型的影响
[0098]
固定md:spi为1:1,单甘脂用量1.0g,芯材比5:1,均质压力50mpa,均质时间10min,进料速率15%,分别选择壁材浓度为2%、4%、6%、8%、10%,制备微囊。分别测定计算微囊的包封率、收率及载药量。由表8可知,随着壁材浓度的增加,微囊的包封率和收率呈先增后减的趋势,当壁材浓度为6%时,包封率和收率最高。
[0099]
表8壁材浓度对微囊的影响
[0100][0101]
2、壁材配比对微囊成型的影响
[0102]
固定壁材浓度为6%,单甘脂用量1.0g,芯材比5:1,均质压力50mpa,均质时间10min,进料速率15%,分别选择md:spi为0.5:1、1:1、1.5:1、2:1、2.5:1,制备微囊。分别测定计算微囊的包封率、收率及载药量。由表9可知,随着壁材浓度的增加,微囊的包封率和收率呈先增后减的趋势,当壁材配比为1:1时,包封率和收率最高。
[0103]
表9壁材配比对微囊的影响
[0104][0105]
3、芯材比对微囊成型的影响
[0106]
固定壁材浓度为6%,md:spi为1:1,单甘脂用量1.0g,均质压力50mpa,均质时间10min,进料速率15%,分别选择芯材比为3:1,4:1、5:1、6:1、7:1,制备微囊。分别测定计算微囊的包封率、收率及载药量。由表10可知,随着芯材比的增加,微囊的包封率先增加然后基本平衡,而收率呈先增后减的趋势,当芯材比为5:1时,收率最高。
[0107]
表10芯材比对微囊的影响
[0108][0109]
4、进料速度对微囊成型的影响
[0110]
固定壁材浓度为6%,md:spi为1:1,芯材比为5:1,单甘脂用量1.0g,均质压力50mpa,均质时间10min,分别选择进料速率为10%、15%、20%、25%、30%,制备微囊。分别测定计算微囊的包封率、收率及载药量。由表11可知,随着芯材比的增加,微囊的包封率和收率呈先增后减的趋势,当进料速率为15%时,包封率和收率最高。
[0111]
表11进料速度对微囊的影响
[0112][0113]
5、微囊制备工艺的正交试验
[0114]
(1)正交试验设计
[0115]
根据前期的单因素实验结果,选取壁材浓度(a),壁材配比(b)和芯材比(c)3个因素,各选择3个水平按l9(34)正交表进行正交试验设计,以包封率、收率和载药量为评价指标。因素水平、正交试验条件及结果见表12,方差分析见表13。由表12可知,各因素对微粉制备工艺的影响顺序为a>c>b。综合包封率和收率的计算结果,确定最佳制备工艺为a3b2c3。即壁材浓度8%,壁材配比md:spi=1:1,芯材比7:1。
[0116]
表12天麻提取物微囊制备工艺正交试验条件及结果
[0117][0118]
表13方差分析
[0119][0120]f0.05
(2,2)=19.00
[0121]
(2)验证试验结果
[0122]
按最佳制备工艺制备三批天麻微囊粉,对预测结果进行验证,实验条件及结果见表14。
[0123]
表14验证试验结果
[0124][0125]
由表13可知,天麻提取物微囊包封率高达到87.48%,收率达到75.96%,载药量达到15.46%。
[0126]
实施例3:天麻提取物微囊的形貌及结构表征
[0127]
1、微囊形貌和粒径
[0128]
取少量天麻提取物微囊粉样品和天麻冻干样品进行真空离子喷金后放入扫描电
镜中进行成像观察,得到扫描电镜图见图6。通过电镜图片观察发现,制备成微囊粉前天麻冻干样品处于无定形的块状结构,大小不一,而天麻提取物微囊呈圆球形,形态稳定,表面光滑圆整,利于粉体的分散。从图7的粒径分布曲线可以看出,在最佳工艺下制备的天麻提取物微囊粒径分布符合正态分布,微囊的累计粒度分布百分数达到0.5时所对应的粒径为4.356μm,说明制备的微囊粒径分布均匀。
[0129]
2、红外光谱表征结果
[0130]
红外光谱法是鉴定化合物种类和化合物结构的重要方法之一,本研究分别称取天麻冻干粉、麦芽糊精、大豆分离蛋白和天麻提取物微囊适量,通过傅里叶变换红外光谱仪在室温下进行测定,扫描范围400-4000cm-1
,光谱图如图8所示。傅里叶变换红外光谱(ftir)图主要用于确认天麻提取物、麦芽糊精和大豆分离蛋白的存在,并研究它们之间可能发生的相互作用。天麻提取物微囊和其辅料在3700~3100cm-1
区间有宽强峰,是o-h键的伸缩振动峰。天麻提取物冻干粉和微囊曲线在1700-1600cm-1
区间为双峰(天麻提取物微囊中为峰1654cm-1
和峰1731cm-1
,天麻提取物冻干粉中为峰1614cm-1
和峰1723cm-1
),是天麻特征峰。天麻提取物微囊曲线中1200-1000cm-1
区间的吸收峰较原料及辅料曲线中该区吸收峰变多变强,是大豆分离蛋白与麦芽糊精共价结合后羟基数目增加所导致。这些结果证明天麻提取物被成功载入微囊中。
[0131]
3、x射线衍射结果
[0132]
天麻冻干粉、麦芽糊精、大豆分离蛋白和天麻提取物微囊的x射线衍射光谱(xrd)图如图9所示。从天麻提取物冻干粉和辅料的结晶结构到微囊的结构发生了明显变化,原料药和辅料为弥散性的衍射峰,而微囊的xrd图中在21.3处出现了新的特征峰,说明原料药与辅料之间发生了弱相关作用,导致了晶型上的变化。
[0133]
4、tg/dsc热分析
[0134]
将天麻冻干粉、麦芽糊精、大豆分离蛋白、和天麻提取物微囊进行tg/dsc测定,结果如图10和图11所示。天麻提取物冻干粉从120℃开始气化,到温度为200℃左右时完全气化。大豆分离蛋白和麦芽糊精从230℃开始气化,到335℃左右气化完全。天麻提取物被和辅料包裹形成微囊后,气化温度提升至190℃,热稳定性增强。
[0135]
5、天麻提取物微囊的热稳定性试验
[0136]
为确定高温条件下天麻提取物在天麻提取物微囊中的稳定性,称取适量天麻提取物微囊和物理混合物各三份分别置于玻璃皿中,在40℃、60℃、80℃的条件下放置10天,分别于第0、1、2、4、6、8、12、20、24、28、32、36、42、54、96、144、240h取样测定天麻提取物微囊中的6种指标成分含量,并绘制热稳定释放曲线,结果见图12,其中a、b、c分别为40℃、60℃、80℃环境下。
[0137]
图12中a显示,24h后物理混合物的相对含量下降加快,54h时天麻提取物微囊中天麻提取物的相对含量为80.37%,而物理混合物中天麻提取物的相对含量则不到70%,到240h时,天麻提取物微囊中天麻提取物相对含量为74.97%,物理混合物中为59.89%。图12中b显示,天麻提取物微囊的相对含量下降曲线与物理混合物的曲线相比更为平缓,36h时天麻提取物微囊中天麻提取物的相对含量为85.97%,而物理混合物中天麻提取物的相对含量为71.04%,到240h时,前者的天麻提取物相对含量为62.76%,后者为51.22%。图12中c显示,天麻提取物微囊和物理混合物中的天麻提取物下降程度均比40℃和60℃时更明显,
到240h时,天麻提取物微囊的天麻提取物相对含量为57.91%,后者为40.76%。
[0138]
综上,在三种温度下,与未处理的物理混合物相比,将天麻提取物包含进微囊中能够显著提高天麻提取物的稳定性,但过高的温度对天麻提取物微囊中的天麻提取物稳定性有一定程度的影响,因此储存时应避免高温条件。
[0139]
6、天麻提取物微囊的体外溶出特性
[0140]
(1)溶出介质的配制
[0141]
为了模拟天麻提取物微囊在人体中的溶出情况,选择人工肠液作为溶出介质。称取磷酸二氢钾3.4g,加入适量纯化水,采用0.1mol/l的氢氧化钠溶液调节ph至6.8;另取5g胰蛋白酶,加入适量纯化水搅拌至完全溶解,将两种溶液混合后加入500ml容量瓶中,加入纯化水定容,即得到人工肠液。
[0142]
(2)微囊溶出度的测定
[0143]
取天麻低温提取物冻干粉200mg及天麻提取物微囊2g,精密称定,加入500ml的人工肠液中,37℃恒温水浴,转速50r/min,分别在0.02、0.05、0.08、0.12、0.25、0.75、1.5、2.0、4.0、6.0、8.0h取样1ml,同时补充人工肠液1ml,按照色谱条件进样测定。按照如下公式计算累积溶出量(q):
[0144][0145]
w表示微囊总质量(mg);dl表示微囊中有效成分的含量;cn表示第n个取样时间点下所取样品中药物的浓度(mg/ml);ci表示第i个取样时间点下所取样品中药物的浓度(mg/ml);v表示溶出介质总体积(ml);vi表示第i个取样时间下的取样体积(ml)。
[0146]
天麻提取物微囊的体外溶出曲线如图13所示,天麻提取物冻干粉10min累积溶出度达90%以上,基本释放完全;天麻提取物微囊1.5h累积溶出80%,2h累积溶出度达到90%以上,前期药物释放速度较快,后期释放速度明显变慢。
[0147]
将天麻提取物微囊的溶出曲线进行模型拟合,由表15可知,微囊中天麻提取物的溶出曲线的零级、higuchi和weibull拟合方程的相关系数分别为0.9361、0.9910和0.9949,表明天麻提取物微囊的体外溶出拟合更符合weibull模型。weibull模型模型在药物溶出曲线评价中应用较多的一种模型,能成功描述各种溶出曲线,其参数对溶出曲线的释放反应灵敏,能使结果的量化更精确,更好地分析和解释数据
]
。天麻提取物微囊的溶出曲线符合weibull模型说明天麻提取物的溶出过程符合扩散释药机制,可以用于预测天麻提取物微囊的体内溶出效果。
[0148]
结合体外溶出曲线及体外溶出模型拟合结果表明,天麻提取物微囊有一定的缓释作用,2h时间基本释放完全,有效解决天麻提取物释放快的问题,可以提高天麻中活性成分的生物利用度。
[0149]
表15微囊中天麻提取物体外溶出拟合结果
[0150]
[0151]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受所述的实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1