4-甲基-5,6-二氢吡喃-2酮在制备NLRP3炎症小体活性抑制剂中的应用
4-甲基-5,6-二氢吡喃-2酮在制备nlrp3炎症小体活性抑制剂中的应用
技术领域
1.本发明涉及nlrp3炎症小体活性抑制剂技术领域,特别涉及4-甲基-5,6-二氢吡喃-2 酮在制备nlrp3炎症小体活性抑制剂中的应用。
背景技术:
2.炎症小体在机体抵抗细菌、真菌和病毒感染的免疫防御和识别自身危险信号中起着 至关重要的作用,它是机体处理大量与应激和损伤相关的致病产物和细胞产物的重要分 子结构。nod样受体蛋白3(nod-like receptor protein 3,nlrp3)炎症小体是迄今为止 研究最多最广泛的炎症小体,它与几种炎症性疾病的发病机制有关,包括克瑞替林相关 的周期性综合征(caps)、阿尔茨海默症、糖尿病、痛风、自身炎症性疾病和动脉粥样 硬化等。nlrp3是一种三元蛋白质,nlrp3炎症小体复合物由一个发挥感知作用的 nlrp3、发挥连接作用的凋亡相关的斑点样蛋白(asc,也被称为pycard)和发挥剪 切作用的caspase-1(半胱天冬酶-1)组成。
3.nlrp3炎症小体激活的经典过程分为两步,包括启动步骤和激活步骤。在第一步启 动步骤中,危险相关分子模式(damps)或细胞受到病原相关分子模式(pamps)等刺 激诱导炎症小体成分nlrp3、caspase-1和白介素1β前体(il-1βpro)的上调表达。在第 二步激活步骤中,nlrp3炎症小体复合物可被多种多样的诱导细胞应激的刺激物完全激 活,nlrp3感知到细胞的应激状态从而被激活。细胞接受刺激物刺激后,nlrp3与衔接 子蛋白asc结合并构建炎症小体复合物并招募caspase-1,导致caspase-1自我切割和激 活。活化的caspase-1剪切il-1β和il-18(白介素18)的前体,导致其活化并释放到细 胞外。
4.il-1β(白介素1β)是早期的炎症标志物,通常被认为是nlrp3信号通路中最重要 的的下游细胞因子,同时许多文献表明il-1β在纤维化的形成过程中也发挥着直接的作 用。il-1β在成纤维细胞中以剂量依赖性方式诱导胶原的表达,il-1β受体缺陷的小鼠在肝 损伤后的纤维化程度减轻,并且il-1β水平升高发生在在纤维化形成之前,hscs可被 il-1β或il-18激活,从而诱导胶原沉积。此外,使用腺病毒在大鼠肺中短暂的过度表达 il-1β促进支气管肺泡灌洗液中tgfβ(转化生长因子β)和pdgf(血小板衍生生长因子) 水平显著升高,il-1β可以诱导得到类似于博来霉素诱导的肺纤维化,以及胶原和纤维连 接蛋白的显著细胞外积聚,且在纤维化的发展中高度依赖于tgf-β和il-17a,这表明 il-1β的刺激也可以升高tgfβ等促纤维化细胞因子的水平。
5.因此,寻找能够抑制nlrp3炎症小体激活的试剂,对于降低炎症并缓解炎症相关疾 病有着重要的意义。
技术实现要素:
6.有鉴于此,本发明目的在于提供4-甲基-5,6-二氢吡喃-2酮在制备nlrp3炎症小体活 性抑制剂中的应用。本发明将4-甲基-5,6-二氢吡喃-2酮用于制备nlrp3炎症小体活性
抑制 剂,具有良好的抑制nlrp3炎症小体激活的功效。
7.为了实现上述发明目的,本发明提供了以下方案:
8.本发明提供了4-甲基-5,6-二氢吡喃-2酮在制备nlrp3炎症小体活性抑制剂中的应 用。
9.优选的,所述nlrp3炎症小体为巨噬细胞的nlrp3炎症小体。
10.本发明提供了4-甲基-5,6-二氢吡喃-2酮为有效成分与药学上可接受的载体组成的组 合物在制备nlrp3炎症小体活性抑制剂中的应用。
11.本发明提供了4-甲基-5,6-二氢吡喃-2酮在制备抗非酒精性脂肪性肝炎药物中的应 用。
12.本发明提供了一种nlrp3炎症小体活性抑制剂,包括活性成分和药学上可接受的载 体,所述活性成分包括4-甲基-5,6-二氢吡喃-2酮。
13.优选的,所述nlrp3炎症小体活性抑制剂的给药剂量为5~200mg/kg。
14.本发明提供了4-甲基-5,6-二氢吡喃-2酮在制备nlrp3炎症小体活性抑制剂中的应 用,本发明发现4-甲基-5,6-二氢吡喃-2酮(化合物c8,cas:2381-87-5)具有良好的抑制 nlrp3炎症小体激活的活性,能够用于制备nlrp3炎症小体活性抑制剂。c8给药后可以 显著降低bdl引起的nlrp3、asc和il-1b等mrna水平的升高,并显著抑制肝组织中 nlrp3、il-1β和asc的蛋白表达水平。c8在bdl大鼠和非酒精性脂肪性肝炎(nash) 小鼠体内表现出良好的降低炎症作用,降低血清转氨酶水平,抑制nlrp3炎症小体的激 活,并且吸收迅速,具有较低的毒性;在体外,c8处理可抑制bmdms细胞中nlrp3炎 症小体的激活,并抑制bmdms细胞培养液上清诱导的lx-2细胞活化。综上,本发明证明 化合物c8是一种安全有效的nlrp3炎症小体活性抑制剂。
附图说明
15.图1是实施例1转录组测序分析动物肝脏组织的基因表达谱变化结果;
16.图2是实施例1表达差异热图;
17.图3是表达差异基因的kegg通路分析图;
18.图4是实施例1中nlrp3炎症小体的主要组成蛋白qrt-pcr验证结果;
19.图5是实施例1中nlrp3炎症小体的主要组成蛋白westernblot验证结果;
20.图6是实施例2中肝脏组织f4/80的免疫组化染色图片;
21.图7是实施例2中肝脏组织f4/80的免疫组化染色结果;
22.图8是实施例2中肝脏组织的f4/80蛋白水平检测;
23.图9是实施例3中蛋白免疫印迹分析细胞和上清中的nlrp3和il-1β蛋白水平;
24.图10是上清处理lx-2细胞后蛋白免疫印迹分析col1a1、α-sma、tgfβ1和il-1β 的表达水平;
25.图11是实施例4中小鼠肝脏病理切片he和天狼星红染色(
×
200)结果;
26.图12是实施例4中天狼星红染色面积统计结果;
27.图13是实施例4中小鼠肝脏组织acta2的mrna表达水平分析结果;
28.图14是实施例4中小鼠肝脏组织α-sma的蛋白表达水平分析结果;
29.图15是实施例4中小鼠肝脏组织col1a1和tgfb1的mrna表达水平分析结果;
30.图16是实施例5中小鼠肝组织中nlrp3、asc和il-1β(17kd)的蛋白水平;
31.图17是实施例5中小鼠肝组织中nlrp3、asc、il-1b、tnf-a和cxcl2的mrna表达 结果;
32.图18是实施例6中c8体内药代动力学分析及初步毒性试验结果;
33.图19是实施例6中h&e染色法对小鼠心脏、肝脏、脾脏、肺和肾脏的病理切片染 色结果;
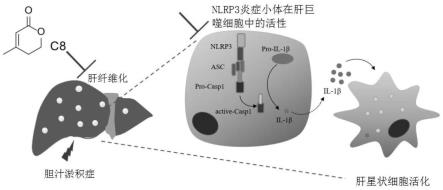
34.图20是c8给药抑制nlrp3炎症小体的激活示意图。
具体实施方式
35.本发明提供了4-甲基-5,6-二氢吡喃-2酮在制备nlrp3炎症小体活性抑制剂中的应 用。在本发明中,所述nlrp3炎症小体优选为巨噬细胞的nlrp3炎症小体,进一步优 选为肝巨噬细胞的nlrp3炎症小体。
36.本发明对所述4-甲基-5,6-二氢吡喃-2在制备nlrp3炎症小体活性抑制剂中的应用 方式没有特殊的要求,根据具体需要,使用本领域技术人员熟知的方法将所述4-甲基-5,6
‑ꢀ
二氢吡喃-2制备成不同剂型的药物即可。在本发明中,所述4-甲基-5,6-二氢吡喃-2酮的 给药剂量优选为5~200mg/kg,更优选为10~100mg/kg,进一步优选为20~50mg/kg。
37.本发明对所述4-甲基-5,6-二氢吡喃-2酮的来源没有特殊的要求,使用市售的4-甲基
ꢀ‑
5,6-二氢吡喃-2酮或自行制备均可。当需自行制备4-甲基-5,6-二氢吡喃-2酮时,所述4-甲 基-5,6-二氢吡喃-2酮的制备方法优选包括以下步骤:
38.在催化剂的作用下,使甲瓦龙酸内酯发生消去反应,得到4-甲基-5,6-二氢吡喃-2酮。
39.在本发明中,所述催化剂优选为一水合对甲苯磺酸;所述甲瓦龙酸内酯与催化剂的 质量比优选为20:1;在本发明中,所述消去反应使用的溶剂优选为甲苯,本发明对所述 溶剂的用量没有特殊的要求,能够将所述甲瓦龙酸内酯和催化剂溶解即可。在本发明中, 所述消去反应的温度优选为反应液的回流温度,时间优选为19h。
40.在本发明中,所述消去反应后还包括对消去反应液的后处理,所述后处理包括以下 步骤:
41.对所述消去反应液依次进行抽滤、硅胶柱层析和洗脱,得到纯净的4-甲基-5,6-二氢 吡喃-2酮。
42.在本发明中,所述抽滤优选为减压抽滤,所述抽滤的时间优选为1h;本发明对所述 硅胶柱层析的方式没有特殊的要求,使用本领域技术人员熟知的硅胶柱层析的方式即可。 在本发明中,所述洗脱的方式优选为石油醚-乙酸乙酯梯度洗脱,所述石油醚-乙酸乙酯的 体积比优选为10:1~2:1。
43.在本发明中,所述消去反应的反应式如式i所示:
44.45.本发明提供了4-甲基-5,6-二氢吡喃-2酮为有效成分与药学上可接受的载体组成的组 合物在制备nlrp3炎症小体活性抑制剂中的应用。
46.本发明对所述载体的种类没有特殊要求,可以包括一种或多种药学上可接受的载 体。
47.在本发明中,所述组合物可以制备成多种剂型,以便于给药。在本发明中,所述组 合物的剂型具体可为肠道外用药制剂,具体的如注射剂、栓剂;所述注射剂可以为溶液、 混悬液或者可注射的干燥粉末(在注射前加入注射水可立即使用);所述注射剂除包括 有效成分外,还优选包括以下载体或助剂:生理上可接受的无菌含水或非水溶液剂、分 散剂、混悬剂、乳剂、合适的含水或非水载体、稀释剂、溶剂或媒介物,具体的如水、 乙醇、多元醇(丙二醇、聚乙二醇、甘油等)、植物油(如橄榄油)和可注射有机酯中 的一种或多种,所述可注射有机酯优选包括油酸乙酯。
48.本发明提供了4-甲基-5,6-二氢吡喃-2酮在制备抗非酒精性脂肪性肝炎药物中的应 用。
49.在本发明中,所述抗非酒精性脂肪性肝炎药物具体优选为口服制剂,所述口服制剂 具体可以为固体剂型或液体剂型;所述固体剂型具体的如片剂、糖衣丸剂、丸剂、散剂、 颗粒剂、胶囊剂或包衣剂,所述液体剂型具体的可为乳剂、溶液剂、混悬剂、糖浆剂或 酏剂。
50.本发明提供了一种nlrp3炎症小体活性抑制剂,包括活性成分和药学上可接受的 载体,所述活性成分包括4-甲基-5,6-二氢吡喃-2酮。
51.本发明对所述药学上可接受的载体没有特殊的要求,使用本领域熟知的药学上可接 受的载体即可。
52.在本发明中,所述nlrp3炎症小体活性抑制剂的给药剂量优选为5~200mg/kg,更 优选为10~100mg/kg。
53.下面结合实施例对本发明提供的4-甲基-5,6-二氢吡喃-2酮在制备nlrp3炎症小体活 性抑制剂中的应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
54.实施例中的实验方法如下:
55.1.lx-2细胞培养
56.(1)细胞的复苏:将冻存的细胞于液氮罐中取出,置于提前打开的40℃水浴锅中快 速溶解,完全溶解后,转移到离心管中,加2ml完全培养基吹打混匀,然后1000rpm, 离心5min,弃上清,细胞沉淀加完全培养基吹打混匀并转移到培养皿中,置于37℃恒温 5%co2细胞培养箱中培养。
57.(2)细胞的传代:待细胞汇合度约为80~90%时进行细胞传代,弃培养基,用pbs 洗一次,在大皿中加1.5ml胰酶,摇晃培养皿底,使胰酶将细胞全部浸润,然后吸走胰 酶,将细胞置于培养箱中消化50s,镜下观察细胞的状态,加培养基终止消化,以1/2的 比例传代或铺板。
58.(3)细胞的冻存:细胞冻存液比例为90%fbs加10%dmso,梯度降温,在负80℃ 冰箱中冷冻24h后转移到液氮中。
59.2.骨髓来源的巨噬细胞(bone marrow-derived macrophages,bmdms)提取分离与培养
60.(1)用10%水合氯醛麻醉小鼠,麻醉后co2处死小鼠,将小鼠后肢的皮毛剥开至足 部,连同褪下的皮毛剪去足部,剪下后腿,放入盛有无菌pbs的培养皿中。镊子夹住腿 骨一端后,用剪刀剔除肌肉,在关节处剪断腿骨。
61.(2)用2ml注射器吸取无血清的1640培养基,将针头刺入骨髓腔,反复冲洗骨髓至 骨髓变白,用枪头将骨髓冲洗液中的细胞重悬,将骨髓冲洗液加入到50ml离心管中,40 μm的滤膜去除冲洗液中其他杂质,然后1000rpm离心5min,沉淀即为骨髓中的细胞。
62.(3)将两个腿骨离心得到的骨髓细胞用16ml含mcsf(50ng/ml),1%p/s和10%fbs 的1640培养基重悬,镜下观察可以看到细胞的形态呈亮而小的圆形,然后分装到8个六孔 板的孔中,置于37℃和5%co2的培养箱中培养3天,在细胞被诱导贴壁前不要换液。
63.镜下观察细胞,贴壁后的bmdm细胞呈长梭形,弃上清,换含有mcsf(50ng/ml)的 1640完全培养基继续培养2天左右即可加药处理,mcsf在细胞培养和加药的全程都是必 需的。
64.3.bmdms细胞诱导与给药
65.为了刺激bmdm细胞中nlrp3炎症小体的激活,将培养基换为无血清培养基,用100 ng/ml lps先预处理细胞3h,然后加10μm尼日利亚菌素刺激细胞0.5h、1h或2h。对于 加药组,化合物c8(20、40、80μm)与lps同时加入细胞中预处理3h,然后加10μm尼日 利亚菌素刺激细胞,收集上清液和细胞裂解液进行westernblot分析。
66.4.胆总管结扎(bile duct ligation,bdl)手术
67.将sprague-dawley雄性大鼠(体重在160-180g)适应性喂养1-2天,将大鼠随机分为 假手术(sham)组、bdl组和c8给药组(bdl-c8),每组8只,同时每组多预留出1只,以防 止动物在手术中意外死亡或者术后因并发症提前死亡。
68.4.1手术
69.手术前一晚,将大鼠禁食,正常饮水。
70.术前给超净台紫外消毒30min,手术器械、缝合针高压灭菌后,浸泡在75%乙醇中, 打开小动物麻醉机,加入足量的异氟烷。
71.待动物完全麻醉后,剃去腹部的毛,用碘伏消毒后,上腹正中开腹,用无菌棉棒抬 高肝缘,拉开十二指肠,找到胆总管,用尖头镊将胆总管与周围的脂肪和结缔组织分离, 在近肝门处和近十二指肠处用缝合线各结扎两道,从两结扎位置中间剪断肝总管,将内 脏恢复原样后滴加1%维生素k(生理盐水配制)止血,并缝合切口,在缝合内皮后撒上 青霉素预防感染,sham组仅开腹,然后缝合。缝合后使动物侧躺,待清醒后自由饮食和 饮水。
72.4.2给药
73.手术后第二天给药,采用灌胃给药,bdl组给生理盐水(ns.)作为对照,连续给药14 天。化合物c8按照50mg/kg的剂量灌胃给药,用生理盐水配制成5mg/ml的混悬液,从手 术后第二天开始称量体重和灌胃给药,给药体积为体重100g/ml。
74.4.3样品收集
75.(1)给药14天后将大鼠移至代谢笼中,禁食12小时,自由饮水,收集期间的尿液。 收集后的尿液静置30min,取上清离心,离心后再取上清,另取一个ep管将尿液稀释10 倍冻存于-80℃备用。
76.腹腔注射10%水合氯醛(3ml/kg)麻醉大鼠,待确认麻醉到足够程度后(眨眼反射
消 失等)从腹正中线开腹,找到腹主动脉取血,血样室温静置30min,3000rpm离心10min, 取上清,冻存于-80℃备用。
77.bdl手术后的大鼠会在肝门下方产生一个大的胆泡,储存了淤积在肝脏中的胆汁, 用2ml注射器胆汁吸出,测量体积,3000rpm离心10min,取上清,另取一个ep管将胆 汁稀释10倍,然后把稀释和未稀释的胆汁都冻存于-80℃备用。
78.再依次取下肝脏、肾脏、回肠,并称肝重和肾重。肝脏最大叶取中间两块分别置于 包埋盒中,泡在10%的福尔马林中固定,剩下的肝叶、肾脏、回肠,切成小块,投于液 氮中,再转移到ep管中,保存在-80℃备用。
79.5.cdaa诱导小鼠形成nash
80.胆碱是形成极低密度脂蛋白(vldl)颗粒膜的主要磷脂合成的底物,缺乏胆碱的饮食 导致vldl无法合成,无法将游离的脂肪转运出肝脏,导致脂肪在肝脏中蓄积,因此喂食 胆碱缺乏饮食的小鼠在不减重的情况下会产生脂肪性肝炎和肝纤维化,形成与人类nash 相似的症状。c57bl/6雄性小鼠(8周龄)以12h/12h的昼夜周期饲养,对照组的饲料为: 胆碱充足的l-氨基酸饲料(choline-supplemented,l-amino acid-defined,csaa,n=7),模型组 和给药组的饲料为胆碱缺乏的l-氨基酸定义饲料(choline-deficient,l-amino acid-definedcdaa,n=7),可自由饮水,c8(100mg/kg,i.p.)从第16周开始给药(cdaa-c8,n=7)维持 4周。在第20周后,采集血清和肝脏样本以供进一步分析。
81.6.生化指标的检测
82.使用日立7180型全自动生化分析仪和中生北控生物科技股份有限公司的生化指标检 测试剂盒,对以下指标进行检测:
83.(1)血清:丙氨酸氨基转移酶(alt)、天门冬氨酸氨基转移酶(ast)、碱性磷酸酶 (alp)、总胆汁酸(tba)、总胆红素(tbil)、总胆固醇(cho)、总甘油三酯(tg)、低密度 脂蛋白胆固醇(ldl-c)、高密度脂蛋白胆固醇(hdl-c)。若检测结果超出范围,用双蒸水 稀释10倍,再次检测。
84.(2)尿样和胆汁:总胆汁酸(tba)和总胆红素(tbil)。
85.7.总rna的提取与纯化
86.(1)样品的处理
87.a:lx2细胞:弃去培养基,按1ml/10cm2的比例加入trizol,静置5min,使细胞充 分裂解,将细胞全部吹打下来,转移到1.5ml的ep管中。
88.b:动物组织:将冻存于-80℃的动物组织称取50~100mg,投于液氮中。在每个玻璃 研磨管(或可用流式管代替)中加入1ml trizol,将组织块取出并投入,用手提式高速 匀浆机在冰水浴中迅速研磨,至无明显组织块。研磨后的组织匀浆在冰上裂解30min, 在4℃离心机中离心15min(12000rpm),离心后吸取上清液转移到新的ep管中。
89.(2)向上述样品中按照每毫升trizol加入200μl的比例加入氯仿,盖紧ep管的盖子, 剧烈振荡15s,室温静置3min。
90.(3)4℃,12000rpm,离心15min,离心后样品分为三层,底层为黄色有机相,上 层为无色水相和一个中间层。rna主要在水相中,水相体积约为所用trizol体积的60%。
91.(4)将上层液体小心转移至新的1.5ml ep管中,大约每管能取出400μl,吸上层液 体时需谨慎操作,不要吸到中间层。
92.(5)使用康为世纪的rna纯化试剂盒纯化rna,首先加入与上层液体等量或稍多的 70%乙醇(无水乙醇:纯水=7:3),上下温和颠倒混匀,室温静置2min然后转移到纯化 柱中。
93.(6)纯化时不需要离心机降温至4℃,常温条件下离心即可(12000rpm,1min),离心 后弃去收集管底的液体,将纯化柱重新放回管中。
94.(7)加入700μl ra1,12000rpm,离心1min,弃去收集管底的液体。
95.(8)加入500μl ra2(首次使用时加入200ml无水乙醇),12000rpm,离心1min, 弃去收集管底的液体,然后重复再加一次ra2并离心。
96.(9)12000rpm,离心2min,离心掉纯化柱中残余的液体。
97.(10)将纯化柱转移到新的1.5ml无rnase的ep管中,开盖10~15min,使残留的乙 醇挥发干净。
98.(11)向吸附膜中间悬空垂直加入65℃预热的无rnase水(细胞的rna量少,可加 20~30μl,动物组织rna量较多,可以加入100~200μl),静置2min,12000rpm,离心 2min收集rna,若浓度过低可将第一次的洗脱液重新加入到吸附柱中进行二次洗脱以增 加rna浓度,每管取出2μl测定浓度与纯度,其余保存于-80℃冰箱。
99.8.逆转录(reverse transcription)pcr合成cdna
100.取1μg总rna,使用roche公司的transcriptor first strand cdna synthesis kit合成 cdna,具体步骤如下:
101.(1)在0.2ml无rnase pcr管中配制如表1所示反应体系:
102.表1反应体系成分
[0103][0104]
将上述组分混匀后,瞬时离心,于65℃变性10min,立即插入冰浴中。
[0105]
(2)向上述体系中加入如下组分,配成20μl表2所示反应体系:
[0106]
表2反应体系组分
[0107][0108][0109]
将所有孔的7μl体系混匀,然后每孔加7μl,混匀后瞬时离心,于55℃反应30min 合成cdna。
[0110]
(3)85℃加热5min失活transcriptor reverse transcriptase,向所得的cdna中
加 入100μl无菌水,混匀后转移到1.5ml离心管中保存于-20℃冰箱或直接用于后续实验。
[0111]
9.real-time pcr实时定量pcr
[0112]
9.1sybr green法
[0113]
以cdna为模板,采用takara公司的sybr rt-pcr试剂盒进行real-time pcr反应, 并以gapdh基因作为内参对照,按下表3配制反应体系(20μl):
[0114]
表3反应体系组分
[0115][0116]
根据样品数和不同的引物,配好以上除cdna外的反应体系,加到real-time pcr用 96孔板中,每孔17μl。再在每孔加入3μl cdna模板,用专用封口膜封住96孔板,将 以上体系混合均匀并离心(1000rpm,5min),离心后,在abi quantstudio 3real-time pcr 实时荧光定量pcr仪中设定反应程序如下:
[0117]
①
95℃5min
[0118]
②
循环40~50次,95℃15s,60℃30s
[0119]
然后立即进行融解曲线程序,单峰的融解曲线证明引物的特异性好,无非特异性扩 增和引物二聚体等干扰实验结果的因素
[0120]
③
95℃15s
[0121]
④
60℃1min
[0122]
⑤
95℃15s
[0123]
反应结束后,根据仪器给出的每个样品的目的基因ct值和内参基因ct值,采用相 对ct值法分析目的基因的相对表达量。
[0124]
sybr green法所用的引物序列如seq id no.1~16所示,具体见表4:
[0125]
表4 sybr green法所用的引物序列
[0126]
[0127][0128]
9.2taqman法
[0129]
使用roche公司的faststartuniversalprobemaster(rox)进行taqman的real-timepcr反应,以所得cdna为模板,并以gapdh基因作为内参对照,按下表5配制反应体系(20μl):
[0130]
表5反应体系组分
[0131][0132]
配好以上除cdna外的反应体系,加到real-timepcr用96孔板中,每孔17μl。再在每孔加入3μlcdna模板,将以上体系混合均匀并离心(1000rpm,5min),离心后,在abiquantstudio3real-timepcr实时荧光定量pcr仪中设定反应程序如下:
[0133]
95℃2min
[0134]
个循环
[0135]
其中,荧光信号值在每个循环的末尾收集,反应结束后,根据仪器给出的每个样品 的目的基因ct值和内参基因ct值,采用相对ct值法分析目的基因和内参基因的表达 量,判断药物对细胞或动物组织的相关基因表达的影响。
[0136]
10.甲醇-氯仿法沉淀细胞培养基上清液中的蛋白
[0137]
(1)将甲醇,氯仿和纯水提前于4度预冷,将培养基上清液转移到15ml离心管中, 然后在1体积培养基上清液中依次加入4倍体积甲醇,1.5倍体积的氯仿和1体积的纯水, 上下剧烈摇匀,然后于4度静置15min,4度12000rpm离心15min,离心后小心吸走上 层甲醇相和底层氯仿相,中间的白色部分即为蛋白。
[0138]
(2)然后加1ml甲醇到管底,将蛋白和甲醇转移到1.5ml ep管中,4度12000rpm 离心5min,弃上清,再加1ml甲醇离心一次,弃上清,用滤纸吸走ep管中残留的甲 醇,晾干20min(尽量挥发干残留的甲醇,否则会影响条带的形态),加适量体积的ripa 溶解蛋白,测浓度后加loadingbuffer煮10min即可。
[0139]
11.蛋白样品的裂解
[0140]
(1)细胞样品:吸出并弃掉原培养基,在孔板或皿中加入pbs小心地洗细胞一次, 加入含有蛋白酶和磷酸酶抑制剂的细胞裂解液,于冰上裂解30min,用细胞刮刀刮下细 胞,收集在1.5ml离心管中。4℃,12000rpm,离心20min,转移上清至新的ep管中。
[0141]
(2)动物组织:将冻存于-80℃冰箱的组织块取出,迅速称取约100mg的组织块, 在每个研磨玻璃管中加入1ml预冷的含有蛋白酶和磷酸酶抑制剂的细胞裂解液,用手提 式高速匀浆机充分研磨匀浆约1min,至管中无明显的组织块变为组织匀浆,放在冰上静 置30min,充分裂解。4℃,12000rpm,离心10min,转移上清至新的ep管中。
[0142]
12.蛋白浓度的测定
[0143]
采用碧云天公司的bca蛋白浓度测定试剂盒(增强型)测定总蛋白浓度,具体步骤 如下:
[0144]
(1)配制标准液:将1.2ml蛋白标准配制液加入到30mg标准蛋白bsa中,充分 溶解,得到25mg/ml的蛋白标准储蓄溶液。取适量25mg/ml蛋白标准,稀释至终浓度 为0.5mg/ml。储蓄液于-20℃长期保存。
[0145]
(2)将0.5mg/ml标准品按0、1、2、4、8、12、16、20的体积加到96孔板的标 准品孔中,加对应体积的双蒸水使总体积为20μl,得到浓度分别为0、0.025、0.05、0.1、 0.2、0.3、0.4、0.5mg/ml的梯度浓度标准品。
[0146]
(3)向每个样品孔中,加入2μl待测样品和18μl双蒸水,得到稀释10倍的样品。
[0147]
(4)根据待测样品数量,配制适量体积的bca工作液。以试剂a:试剂b=50:1 (v/v)的比例配制,充分混匀。向每个标准孔和样品孔中,各加入200μl bca工作液, 在培养板振荡器上振荡混匀约1min,放在37℃孵育箱中孵育30min。
[0148]
(5)在酶标仪波长560nm处测量各孔od值,以标准品的蛋白浓度为横坐标,od 值为纵坐标,绘制标准曲线,根据标准曲线计算待测样品的蛋白浓度。
[0149]
(6)按照1.0-1.5μg/μl浓度,计算相同蛋白总量时各待测样品的体积,余下的体积 用细胞裂解液补齐,将不同浓度的蛋白调整为相同的浓度,加入5
×
loading buffer,使 蛋
白样品终浓度变为1
×
。
[0150]
(7)于沸水浴或95℃金属浴中加热10min,使蛋白变性,加热后立即置于冰上冷 却,使水蒸气凝结,保持终体积不变,冷却后瞬时离心。混匀后即可上样,进行蛋白电 泳,其余样品保存于-20℃冰箱。
[0151]
13.聚丙烯酰胺(sds-page)凝胶的制备
[0152]
a:将厚玻板和薄玻板洗净,晾干备用。首先倒入分离胶,用双蒸水液封,可看到胶 和水之前有明显分界线,分离胶凝后,倒掉上层水,并且用纸巾吸干剩余水分,倒入浓 缩胶,并插入梳子。
[0153]
b:按照表6、表7分别配制分离胶和积层胶:
[0154]
表6分离胶组分
[0155][0156]
表7积层胶组分
[0157][0158]
14.聚丙烯酰胺(sds-page)凝胶电泳及蛋白免疫印迹
[0159]
(1)将凝胶板安装在电泳架上,向凝胶板之间和电泳槽中加入适量的1
×
蛋白电泳缓 冲液,其中凝胶板之间的电泳液要加满。拔掉梳子,加样。一般每孔加入细胞蛋白样品 5-20μg,动物组织样品10-50μg,marker孔中加入预染蛋白marker。接通电源,调节电 压。先在80v电压下电泳,待蛋白完全进入分离胶后,换为120v电压继续电泳,至合 适位置停止电
泳。
[0160]
(2)预处理pvdf膜:提前将裁剪好的pvdf膜,浸泡在甲醇中15秒进行活化,然 后浸泡1
×
转膜缓冲液中。可在左上角或右上角剪去一角,作为标记,以区分膜的正反面 和顺序。
[0161]
(3)将凝胶从板上取下,切除浓缩胶和溴酚蓝前沿部分,浸泡在1
×
转膜缓冲液中平 衡。安装凝胶和pvdf膜,夹板白色透明一侧置于底端,依次放上海绵、滤纸、pvdf 膜、凝胶、滤纸、海绵,再将夹板黑色侧压上,扣紧。组装的过程中,每层都要尽量避 免有气泡,尤其是pvdf膜和凝胶之间。
[0162]
(4)调节电流,180ma恒流转膜1.5h,或调节电压90v恒压,转膜1h-1.5h。此条件 根据目的蛋白分子大小适当调整,分子小的时间要适当缩短。封闭:转膜结束后,取出 pvdf膜,放入5%脱脂牛奶中,室温封闭1-2h。
[0163]
(5)一抗孵育:根据预染marker所标注分子量大小和待测目的条带的大小将膜裁开, 宁多勿少。用1%bsa或者一抗稀释液配制一抗,一抗稀释比例按说明书。准备好抗体 和膜后,将膜放入杂交袋中,再加入相应的一抗,于4℃摇床孵育过夜。
[0164]
(6)漂洗:取出膜条,用1
×
pbs-t漂洗三次,每次10min。
[0165]
(7)二抗孵育:提前准备好二抗,用1%bsa配制,抗体浓度按说明书配制。将膜 放入二抗孵育盒中,加入相应的二抗,于室温孵育2h。
[0166]
(8)漂洗:从杂交袋中取出膜条,用1
×
pbs-t漂洗三次,每次10min。
[0167]
(9)ecl显色曝光:配制新鲜的增强型hrp底物化学发光液(ecl),将底物与缓 冲液按1:1(v/v)的比例混合,均匀的滴加到膜条上,在凝胶成像仪中曝光显色。
[0168]
(10)结果分析:保存曝光图片,用image j处理,与内参比较后,统计蛋白表达变 化情况。
[0169]
15.初步急性毒性实验
[0170]
急性毒性实验是24h内给药一次或2次(间隔6~8h),观察受试动物接受过量的药物 产生的急性毒性反应,可以为其安全性或剂量选择提供参考和依据。本实验中采取单次 口服固定剂量法,选取的剂量为1000和2000mg/kg。将8周龄balb/c小鼠分为3组,每组 3只雌性和3只雄性小鼠,适应性饲养几天,给药之前禁食约8h,然后分别给予小鼠口服 生理盐水,或c8(1000或2000mg/kg,p.o.)。观察小鼠状态14天,记录体重,然后处死小 鼠,收集心、肝、脾、肺和肾样本进行苏木精和伊红(h&e)染色分析。
[0171]
实施例1 c8抑制肝脏中nlrp3炎症小体的表达和活化
[0172]
首先,提取纯化bdl大鼠肝脏组织的总rna并进行转录组测序分析,将表达倍数减 少或增加2倍及以上的定义为显著变化。转录组测序分析动物肝脏组织的基因表达谱变化 (n=3)如图1所示。由图1可以看出,与sham大鼠相比,bdl大鼠肝组织中共检测到2088个 显著下调基因和4933个显著上调基因;与bdl大鼠相比,bdl-c8组大鼠肝组织中5226个 基因表达显著下调,2631个基因表达显著上调;在两个比较组中都有显著改变的有2577 个基因。
[0173]
表达差异热图如图2所示,图2中,红色表示高表达,蓝色表示低表达。表达差异基 因的kegg通路分析图如图3所示。kegg通路富集分析显示,这2577个基因在脂肪酸代 谢、非酒精性肝病和炎症相关信号通路中富集。在这些途径中,nlrp3信号通路是与胆 汁淤积引
0.5h后含有活性il-1β的bmdms培养基以诱导人肝星状细胞系lx-2活化,结果图图10所 示。图10为图9中(c)条件下获得的上清处理lx-2细胞后蛋白免疫印迹分析col1a1、 α-sma、tgfβ1和il-1β的表达水平;蛋白水平用内参gapdh校准,n=3,*p《0.05vs.对照 组;#p《0.05vs.尼日利亚菌素(10μm)0.5小时处理组,$p《0.05vs.尼日利亚菌素(10μm)1 小时处理组。
[0186]
图10结果显示由il-1β诱导升高的纤维化标志物col1a1和α-sma在c8给药后显著降 低。以上结果表明,c8给药抑制了bmdms中nlrp3炎症小体的激活,并且抑制由nlrp3 激活的bmdms释放的il-1β诱导的hscs活化,因此c8是通过抑制肝脏中nlrp3炎症小体 的激活来发挥抗肝纤维化和减轻炎症反应的作用。
[0187]
实施例4 c8给药减轻cdaa饮食诱导的nash小鼠的肝损伤
[0188]
在确认了c8对bdl大鼠的保护作用后,本发明在cdaa诱导的nash小鼠中进一在对 c8的抗肝纤维化药效进行了验证。nafld是肝脏因脂质(甘油三酯、游离脂肪酸、胆固 醇、和神经酰胺等)在肝细胞内过度积聚导致的慢性肝病。近年来nafld等代谢性肝病 的发病率明显上升,据统计,nafld的全球发病率约为25%,nafld包括两种不同的情 况:1)出现脂肪变性或伴有轻度小叶炎症的脂肪变性;2)非酒精性脂肪性肝炎 (non-alcoholic steatohepatitis,nash),出现不同程度的纤维化、肝硬化和hcc,nash患 者约占nafld患者总数的20%。在nash肝硬化患者中,hcc的估计年发病率在0.5%到 2.6%之间,非肝硬化nafld患者的hcc发病率较低,约为每1000患者年0.1至1.3例,尽 管nafld相关hcc的发病率低于其他病因如丙型肝炎引起的的hcc,但nafld患者的 群体较大,nafld相关的hcc仍然会导致沉重的健康负担。极低密度脂蛋白(vldl)颗粒 可以将肝脏甘油三酯(triglycerides,tg)、胆固醇(cholesterol,cho)和载脂蛋白从肝脏输出 到外周组织,而胆碱是合成形成vldl颗粒膜必需的磷脂酰胆碱的底物,胆碱的缺乏会导 致vldl减少,tg无法转运出肝脏,脂肪在肝脏中大量积累。胆碱缺乏的l-氨基酸饮食 (choline-deficient,l-amino acid-defined diet,cdaa)饲养的小鼠在体重不减轻的情况下会 自发产生脂肪性肝炎和肝纤维化,与人类的nash相似。
[0189]
本发明对c57bl/6雄性小鼠给予cdaa饮食20周诱导nash,以胆碱足量的l-氨基酸 饮食(choline-supplemented,l-amino acid-defined diet,csaa)为对照,c8给药组 (cdaa-c8)在第16周开始腹腔给药进行治疗,给药4周。
[0190]
小鼠肝脏病理切片he和天狼星红染色(
×
200)结果如图11所示。天狼星红染色面积统 计采用imagej软件分析结果如图12所示;图12中,数据用平均值
±
sd值表示,针对gapdh 标准化,*p《0.05,**p《0.01vs.csaa组;#p《0.05,##p《0.01vs.cdaa组。
[0191]
小鼠肝脏组织acta2的mrna表达水平分析如图13所示;小鼠肝脏组织α-sma的蛋白 表达水平分析如图14所示;小鼠肝脏组织col1a1和tgfb1的mrna表达水平分析如图15所 示。
[0192]
血清生化指标的的结果显示,与cdaa喂养的小鼠相比,c8给药的小鼠血清中的alp 平均值显著降低。肝组织切片天狼星红(sirius red)和he染色的结果表明,cdaa饮食诱 导的小鼠肝细胞气球样变性和胶原纤维沉积显著增加,而c8给药后有效改善小鼠的肝纤 维化水平。western blot和qrt-pcr的结果显示,c8给药可抑制肝组织中纤维化标志物 α-sma的mrna和蛋白质表达水平,降低col1a1和tgfb1的mrna平均值但无显著性差异, 但这些结果表明,c8处理可减轻nash小鼠的肝损伤和纤维化程度。
[0193]
实施例5 c8给药抑制nash小鼠中nlrp3的激活
[0194]
nlrp3炎症小体的激活在cdaa诱导的nash小鼠的肝纤维化发展过程中发挥了重 要的作用,阻断其激活或敲除nlrp3可显著减少nash小鼠的肝脏炎症水平和纤维化程 度。
[0195]
检测nash小鼠肝组织中nlrp3信号通路及下游炎症因子的表达水平。小鼠肝组织 中nlrp3、asc和il-1β(17kd)的蛋白水平如图16所示;小鼠肝组织中nlrp3、asc、il-1b、 tnf-a和cxcl2的mrna表达如图17所示;图17中数据以平均值
±
标准差表示,与gapdh标 准化,n=7,*p《0.05,**p《0.01vs.csaa组;#p《0.05,##p《0.01vs.cdaa组。
[0196]
western blot和qrt-pcr的结果表明,nash小鼠肝组织nlrp3、il-1β和asc的蛋白 表达以及nlrp3、asc、il-1b、tnf-a和cxcl2的mrna水平显著升高,这表明在cdaa饮食 导致的肝损伤过程中nlrp3信号通路的激活,而在c8给药可以显著降低这些指标的表达 水平,即c8处理可以降低cdaa诱导的nash小鼠中肝脏nlrp3的表达和激活。
[0197]
实施例6 c8在小鼠中的药代动力学及初步急性毒性实验
[0198]
药代动力学和毒性是化合物成药性的重要因素,在之前的动物实验中c8的给药方式 为腹腔注射100mg/kg,实验中未观察到c8给药对大鼠和小鼠的毒性。接下来对c8的药代 动力学及初步急性毒性进行考察。药物动力学在sd大鼠中进行,分别口服给药(50mg/kg) 或静脉注射给药(50mg/kg),在不同的时间点取血放置于肝素钠抗凝管中然后离心获得血 浆,通过液相二级质谱(lc-ms/ms)检测给药后不同时间点血浆中的化合物含量。结果如 表8所示。
[0199]
表8口服或静脉给药后化合物c8的药代动力学参数
[0200][0201][0202]
注:auc,曲线下面积;t
1/2
,消除半衰期;c
max
,最大血浆浓度;tmax,达到最大 血浆浓度的时间;mrt,平均停留时间。
[0203]
初步的急性毒性实验则在balb/c小鼠中进行,采用单次口服固定剂量方法,小鼠分 为三组(ns.,c8-1000mg/kg,c8-2000 mg/kg),每组6只雌雄各半,在给药后及之后的14天 中观察小鼠的生理状态和行为状态有无异常,每天称量体重并记录。
[0204]
其中,c8体内药代动力学分析及初步毒性试验结果如图18所示。图18中,(a)为 口服(50mg/kg)后c8的血浆浓度,(b)为静脉注射(2mg/kg)后c8的血浆浓度;(c)、 (d)为口服c81000 mg/kg、c82000 mg/kg或ns的小鼠体重。h&e染色法对小鼠心脏、 肝脏、脾脏、肺和肾脏的病理切片染色结果如图19所示。
[0205]
结果显示,口服(p.o.)或静脉注射(i.v.)后,分别约在0.33h和0.08h时达到了最高血药 浓度(c
max
,17918.97
±
1436.43ng/ml,1889.87
±
21.17ng/ml),说明c8可以很快的被吸收进 入血液中,其消除半衰期(t
1/2
)分别约为1.07h和0.75h,在给药4h和8h后几乎检测不到血 液中的化合物含量,表明虽然c8能够被快速吸收,但是其被代谢清除的速率也很快,在 接下来的体内研究中,可以降低给药剂量,增加给药次数,从而稳定血药浓度,得到更 好的治疗效果。
[0206]
在本次实验中,小鼠口服c8后表现出呼吸急促,行动减少或静止休息的状态,约在 半小时至一小时后逐渐恢复正常行动状态,小鼠的体重在14天内正常波动,与ns.组无显 著差异,但在口服2000mg/kg的小鼠中有2只死亡(雌雄各一只);各组小鼠的心肝脾肺 肾h&e染色结果显示心肌纤维和肝脏细胞无肿胀,无炎症浸润和纤维化产生,肺泡和细 支气管无扩张或塌陷,脾脏中细胞形态正常,肾脏中肾小球、近端小管和远端小管的轮 廓清晰,数量正常,以上结果表明c8给药后对各个脏器无明显毒性。
[0207]
以上实施例结果表明,在胆汁淤积大鼠和nash小鼠中,c8给药后,nlrp3及衔接 子蛋白asc和最重要的下游细胞因子il-1β的蛋白水平降低,其他nlrp3信号通路相关的 细胞因子如tnfα和cxcl2的mrna表达也显著降低,表明c8给药抑制了肝脏炎症水平,这 也与h&e染色病理切片中炎症和坏死评分降低的结果一致。在肝脏损伤后的炎症过程中, 肝脏常驻巨噬细胞kcs的数量因为消耗而减少,并且被骨髓来源的单核细胞取代,然后 这些单核细胞分化为新的肝巨噬细胞,因此文献中常用骨髓来源的巨噬细胞来探讨肝脏 中nlrp3炎症小体的激活。nlrp3可以被各种引起细胞压力过大的刺激物所激活,其中 最常用的刺激条件是lps预处理3~4h后,再加尼日利亚菌素或atp或尿酸单钠(msu)晶体 刺激nlrp3活化。因此本发明分离了balb/c小鼠的bmdms,并以lps和尼日利亚菌素 诱导。在bmdm中,lps和尼日利亚菌素诱导的nlrp3、il-1βpro和分泌到细胞培养液 中的il-1β增加,而c8处理组的细胞中其增加被抑制,表明c8处理抑制nlrp3的活化。此 外,免疫组化的结果显示bdl大鼠肝组织中巨噬细胞表面标志物f4/80的增加在c8给药组 也明显降低,表明c8给药减少巨噬细胞的浸润,并且是通过抑制nlrp3的激活抑制炎症 反应。
[0208]
纵向研究表明,在nafld患者中,与无纤维化(0期)的患者相比,死亡率风险和 肝脏相关发病率的风险随着纤维化阶段的增加而增加(0期vs4期),肝脏相关预后也主 要与肝纤维化的程度有关,肝纤维化程度高的nafld患者的预后更差。而巨噬细胞中 nlrp3的激活是hscs活化的重要原因,本发明实验结果表明含有成熟il-1β的bmdms培 养基上清液可诱导lx-2细胞活化,诱导纤维化标志物col1a1和α-sma等的表达增加, 而c8处理可抑制il-1β诱导的lx-2活化。综上,本发明实验表明c8给药抑制nlrp3炎症 小体的激活,减少促纤维化细胞因子的释放,并直接抑制tgfβ1和il-1β诱导的hsc激活, c8给药抑制nlrp3炎症小体的激活示意图如图20所示。
[0209]
许多研究表明,caspase1在nlrp3信号通路的激活中起着关键作用,然而,c8给药 后,动物肝组织和bmdm细胞均未观察到caspase1的明显变化。虽然c8对caspase1没有明 显
的升高或降低作用,但近期的文献表明caspase1本身在nlrp3的激活过程中并不发挥 决定性作用,敲除nlrp3或il-1β可以有效的减少胆管闭锁引起的肝脏损伤,改善生存率, 而只敲除caspase1对肝脏没有保护作用,不能减少肝脏的损伤,对生存率也没有改善。 考虑到c8对nlrp3和il-1pro蛋白表达的抑制作用,我们认为c8主要在“第一阶段”发挥作 用,即抑制nlrp3以及il-1βpro的表达水平后,caspase1可以剪切的il-1βpro减少,最终 导致成熟il-1β的细胞外分泌减少。
[0210]
本发明研究结果表明c8具有较好的的生物利用度和安全性,在1000mg/kg的浓度下, 未检测到c8的毒性作用,并且在给药后的短时间内,血浆中c8的含量达到很高的浓度, 这表明c8可以在体内大量吸收。然而,c8在体内的代谢也很快,口服8h或腹腔注射4h 血液中的化合物水平几乎已经检测不到,因此减少给药剂量和每天多次给药可能会进一 步改善其治疗效果。
[0211]
本发明阐明了化合物c8治疗肝纤维化的作用机制,并初步探讨了其急性毒性和药代 动力学特征。在体内,化合物c8在bdl大鼠和nash小鼠体内表现出良好的抗肝纤维化 和降低炎症作用,降低血清转氨酶水平,抑制nlrp3炎症小体的激活,并且吸收迅速, 具有较低的毒性;在体外,c8处理可抑制tgfβ1和il-1β诱导的lx-2细胞活化,并抑制了 bmdms细胞中nlrp3炎症小体的激活,综上,本发明证明化合物c8是一种安全有效的 nlrp3炎症小体活性抑制剂。
[0212]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员 来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也 应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1