一种基于十进制的DNA存储编码方法、设备及可读存储介质与流程
一种基于十进制的dna存储编码方法、设备及可读存储介质
技术领域
1.本发明涉及一种基于十进制的dna存储编码方法、设备及可读存储介质,属于生物技术与信息技术领域。
背景技术:
2.dna存储是指使用dna当作介质来存储信息数据。dna分子极其稳定,不需要额外消耗能量维护,在低温干燥的环境下能够保存百万年之久,并且极其微小(单个碱基加上磷酸核糖骨架,合计仅三四十个原子),存储密度极高。理论上一克dna可以保存人类迄今为止拍摄的所有电影,或者人类有史以来的所有书籍和绘画等资料信息。这两个优点远远超过目前所有的其它信息存储介质,如纸张、光盘、磁盘、磁带等。dna分子不会依赖于特定的读取设备,这点也不同于目前的电子设备,比如三十年前最流行的软盘,由于读取设备不再生产使得数据的读取非常麻烦。但dna是地球几乎所有生物的遗传物质,无论未来科技如何发展,人类总会有各种方法读取dna数据,仪器设备如何改变都不会影响对dna序列信息的读取。作为存储介质,dna的缺点是不能任意修改数据,读写时间慢,成本很高。尽管如此,使用dna来长期备份保存档案资料等惰性信息,即很少被使用却很重要的高价值数据信息资料,依然具有广阔的前景。
3.dna存储的一个重要研究方向是编码方法,即如何将二进制数据高效地转换为碱基序列,将二进制数据存储在含有四种碱基的dna序列中。但在dna序列中,要求单碱基的重复要尽可能少,gc和at含量比要均匀(介于大约40%-60%之间),这样才不会因为双链结合过于牢固,对dna的合成和复制带来困难而影响dna的测序。直接对二进制序列转换的方法会导致生成的dna序列达不到上述要求,因此转换时需要尽量避免这种情况的出现。
4.经过十多年的发展和研究,在dna存储编码方面比较代表性的编码方法有美国哈佛大学提出的二进制,欧洲生物信息中心提出的三进制,美国哥伦比亚大学提出的喷泉码四进制、美国华盛顿大学的混合进制等几种方法,这几种方法采取了不同的策略解决序列限制的问题。
5.二进制的方法在将二进制数据转换为dna序列时,每一个比特用一个碱基表示。单个比特有0和1两种,其中0用a或者c表示,1用g或者t表示。或者说,a和g都可以表示0,c和t都可以表示1。这样就可以调整序列而不会改变其编码的信息,从而避免出现gc含量不均匀或者重复序列的问题。
6.三进制方法则首先将二进制数据按照8比特256个字符(0~255),使用哈夫曼方法将其压缩到243个字符,然后用5位三进制数表示,因为后者同样是243个字符(35)。再将5位的三进制字符直接转换成5碱基的dna序列,转换过程要求相邻的两个碱基不能相同,因此每个位置的碱基必须和前面相邻碱基不同,所以只有其它三种碱基选择,分别对应三进制的0,1,2这三个数字。根据预先设定的碱基转换表进行,每个碱基具体表示0,1还是2取决于前面碱基是什么,因此需要设定一个起初碱基即可进行转换。
7.四进制则是改造成熟的喷泉码,首先将二进制数据按照32字节(256比特长度)分
段,再将每一段32字节通过ruby转换生成新的32字节二进制数据,这个过程可逆,并且可以有很多种不同的随机转换结果,然后把新的二进制数据按照每个碱基对应2个比特直接转换,并判断得到的dna序列是否符合序列限制的要求,如果不符合要求则重新开始ruby转换,直到获得的dna序列符合序列限制的要求为止。
8.混合进制则是首先将6比特数据,按照每个碱基2个比特转变成3个碱基,再添加一个碱基来防止多个三碱基码字排列可能出现的重复序列或高gc含量。如ggg后面添加一个a变成ggga,aaa后面添加一个c变成aaac,这样也和三进制一样需要预先设定一个6比特到四碱基转换表,按照这个表将二进制数据转换成dna序列。
9.国内也有学术机构和企业进行相关研究,但一般都是不考虑序列限制直接转换。此外,国内申请的dna存储专利有一部分是使用长双链dna分子,比如超过1000bp(bp,碱基对)或更长来编码并存储信息,这一方向实际上已经基本被抛弃。原因有二,一是上述方法同样可以使用长链dna分子;二是所有的长链dna分子也都是要先合成短链寡核苷酸,然后再用分子生物学方法拼接获得长链分子。因此使用长双链dna增加了步骤,成本比直接使用短链寡核苷酸高百倍以上,但却没有任何实质的突出优点,也没有避开显著的困难。dna分子的信息潜力极其巨大,比如200个碱基的长度,就算两端引物去除合计50碱基,剩下的150碱基用于编码,其碱基的序列种类合计也有4
150
个,这个数字超过10
90
,远远高于目前已知宇宙的基本粒子总和的数量(大约不到10
70
),而目前人类每年生产的所有电子存储设备总容量大约在10
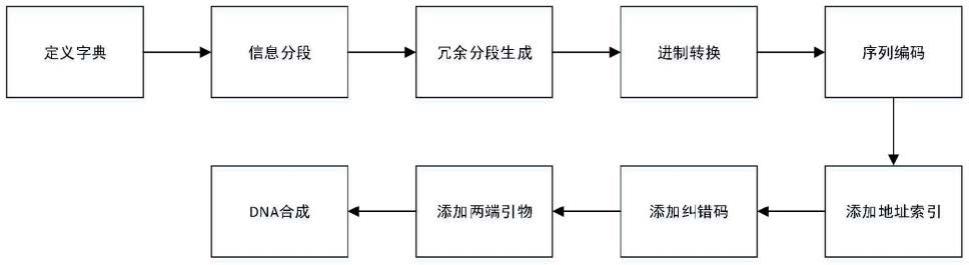
21
这个级别。即使考虑到碱基序列平衡,其潜力也远远足够人类存储信息的需求,因此无需逐个将短片段拼接成长链dna存储信息。
10.中国的华大基因研究团队在2022年4月发表文章,提出新的编码方式阴阳码,具有较高密度,其原理本质上也是和喷泉码一样,将二进制直接转换成碱基,只是使用了伪随机的方式,这种转换需要随机挑选两段相同长度的二进制数据逐位比较,按照两套不同的编码规则将两段数据同一位置的各自一个比特,合计两个比特,再结合dna序列的前面一个碱基具体是什么碱基,使用一个确定的碱基记录这两个比特,最终将两段二进制数据存入相同长度的一段碱基序列,然后评估这段碱基序列是否符合序列限制的要求,如果不符合则丢弃重新随机挑选两段二进制数据重新来一遍,直到碱基序列符合要求。该方法声称使用短寡核苷酸存储数据时,质量密度比喷泉码高10倍,但根据其实验条件看,原因可能是其对比的喷泉码使用仅7%的序列冗余,而阴阳码却使用20%的序列冗余造成的,并且喷泉码模拟测试最低10倍测序深度即可解码获得全部数据,阴阳码即使百倍深度覆盖测序,也未能百分百解码出全部数据。其编码的总数据量也较小,大约仅0.5mb左右,而喷泉码作者编码了2m多数据,混合进制作者编码了200m数据,明显不是一个数量级。方法还同时使用阴阳码编码了一段信息,通过逐级拼接合成约54kb(kb,千碱基对)的长链存储这段信息,并将克隆到酿酒酵母基因组中,但方法也未能说明用长链dna存储数据有什么重要意义,或者有什么其它方法不能这么做的理由。
11.dna编码需要解决的另一问题是,短寡核苷酸上所能够存储的信息有限(一两百个比特),即二三十个字节。那么存储大量数据就必然需要极多的不同序列的dna分子,这些各自携带有限信息的海量dna分子如何排序的问题也需要解决。目前的方法有两个思路,三进制采用片段重叠覆盖的方法,二进制、喷泉码、混合进制和阴阳码采用地址索引的方法。
12.重叠覆盖即多倍分子序列两端重叠,比如一个字符串abcdefgh要分段保存,每段4
个字母,那么片段分别是abcd,bcde,cdef,defg,efgh,每一个片段都和其它的有重叠,所以能够排序。三进制使用100碱基长度片段,每个都分别有75碱基和前后相邻片段重叠,总共使用4倍覆盖的序列保存信息,因此其实际存储密度大大减小。重叠覆盖也需要高倍的测序深度来纠正随机错误,并保证测序结果必须覆盖所有信息区域。这个思路的优点是单组dna容量没有明显上限,缺点是单碱基信息密度太低,几乎做不到百分百正确恢复数据。
13.地址索引是在dna上单独留出一小段序列用于该dna片段的地址索引。dna喷泉码使用16碱基序列,混合进制编码使用14碱基长度序列,来当作地址索引给每条序列精确定位。按照作者的索引数字产生方法,16碱基最多能够获得千万个左右有效索引。使用地址索引,片段之间不需要有任何重叠。由于实际上在合成dna片段时,合成量并不均匀,获得的寡核苷酸的绝对量有多有少,还会出现部分寡核苷酸片段的丢失。这两个编码方法分别使用喷泉码和rs纠错码产生冗余片段,保证在一般测序深度下,即使丢失部分片段的信息,也都能够正确恢复全部数据,因此冗余设计对dna存储的编码方法非常重要。
14.重复覆盖方法不需要额外单独保存地址索引信息来记录片段之间的先后顺序关系,而地址索引方法需要将这种关系记录在电脑系统中,由于索引本身也要考虑到序列限制,一般是使用随机数字转变成序列,因此可能需要将所有的索引数据一一记录,以备恢复数据时参考比对。这些数据需要保存和维护,万一这些地址索引数据丢失,dna中的数据将不可恢复,这对于信息在dna中的永久保存是个致命缺陷。且由简单计算可知,一个序列中地址索引的数据量至少是其保存数据量的十分之一,dna存储比电子设备存储价格要高百万倍以上,一旦索引信息必须仍然存在电子设备中,那么将信息存储在dna中而不是直接存储在电子设备中就没有实际的价值和意义。如果使用dna保存备份人类文明的精华信息,如文学,历史,音乐,绘画等数据,这些数据可能会在千年,万年或者百万年后被使用,那么地址索引数据的维护和保存就会成为极其具有挑战性的问题。
15.一般认为,单组dna的容量可达到tb级(10
12
bytes)才能满足实际应用的需求。混合进制编码方法文章里,详细分析并筛选出超过6千条引物,它们可以任意组合进行多重pcr扩增,可以依靠不同的引物组合将单个寡核苷酸池容量提高到tb以上,并使用了超过千万条150碱基长度的dna片段(oligo)存储多个文件,合计200m数据。存储时对不同的文件使用不同的引物组合以便直接读取文件,使用不同引物组合尽管可以提高单组dna的数据容量,但也是目前的无奈之举,一旦存储的单组数据dna的容量足够大,就会有后遗症问题。比如用不同的引物组合,使得单组dna的容量达到了tb级别,那多组dna之间如果有了污染就将会影响数据的读出。假如不依靠引物组合就能够使得单组dna容量达到tb级别,那么不同组的dna即可以使用不同引物组合,也能够在dna之间出现污染时进行区别。
技术实现要素:
16.发明要解决的技术问题
17.本发明针对现有dna存储编码方法的问题,提出一种基于十进制的dna存储编码方法,该编码方法具有适合的信息密度,在不依赖引物组合的前提下,彻底解除单组dna的容量上限,并且地址索引无需在电脑系统中保存维护。
18.技术方案
19.为达到上述目的,本发明提供的技术方案为:
20.一种基于十进制的dna存储编码方法,包括如下步骤:
21.步骤1,字典定义:使用非重复的双碱基码字对应0~9十个阿拉伯数字创建字典,采用ac,ca,ag,ga,tc,ct,tg,gt编码0~7;at,ta编码8和9,gc和cg作为8和9两个数字的替补码。
22.步骤2,信息分段:将待存储二进制信息按照n比特的倍数进行分段。
23.步骤3,生成冗余分段:按照一定的冗余比例为一组分段后的信息生成若干冗余分段,冗余分段的长度为与上一步信息分段长度相同,冗余分段的每个字节由该组内所有分段信息对应字节根据rs纠错码生成规则随机生成,冗余分段与原信息分段共同组成一个基本纠错单元,即一个码块。
24.步骤4,进制转换:将每个分段信息的二进制序列转换为十进制序列,
25.步骤5,序列编码:根据字典使用双碱基dna码字替换十进制数字序列。
26.步骤6,添加地址索引:将上一步骤所得到的每一条碱基dna序列使用一定长度的十进制数字串记录该序列的精确位置,根据字典使用双碱基dna码字对上述十进制地址生成对应碱基长度的dna序列,将地址碱基序列添加到序列编码所得碱基序列作为该碱基序列的地址索引。
27.步骤7,添加纠错码:在上一步生成的碱基长度序列尾部添加一定碱基长度的纠错码,纠错码是汉明码或者rs纠错码。
28.步骤8,添加两端引物:在上一步所得碱基序列首尾两端各添加一定碱基长度的引物。
29.步骤9,dna合成:将前述步骤所获得的dna序列以阵列芯片的方法进行合成,并存储至相应的容器中。
30.进一步地,步骤2中将待存储二进制信息按照n比特进行分段,n为13,3或6的倍数。
31.进一步地,步骤7中纠错码为rs纠错码或汉明码。
32.进一步地,步骤7中汉明码为2进制汉明码或4进制汉明码。
33.一种基于十进制的dna存储编码设备,包括:
34.存储器,用于存储计算机程序;
35.处理器,用于执行所述计算机程序时实现如权利要求1-4任一项所述基于十进制的dna存储编码方法的步骤。
36.一种计算机可读存储介质,所述计算机可读存储介质上存储有计算机程序,所述计算机程序被处理器执行时实现如前述基于十进制的dna存储编码方法的步骤。
37.有益效果
38.本发明方法采用了多种纠错码,能够更加有效地对存储序列进行纠错并恢复所有存入信息;
39.本发明方法较现有dna编码方法相比在保证dna存储密度的同时保证了存储数据的完整恢复;
40.本发明方法能够突破单组dna容量的限制,满足大容量存储要求。
附图说明
41.图1为本发明存储编码方法的步骤图。
具体实施方式
42.为进一步了解本发明的内容,结合附图和具体实施方式对本发明作详细描述。
43.如图1所示,本发明存储编码方法包括如下步骤:
44.定义字典:双碱基码字是指两个碱基排列构成的dna序列码,碱基共有4种,因此双碱基码字一共有16种,将双碱基相同的4个(aa,cc,gg,tt)去掉后剩下的10个(ac,ca,ag,ga,tc,ct,tg,gt,at,ta)分别编码0到9这十个阿拉伯数字,另外gc和cg作为替补码字分别编码8和9。
45.字典的简洁规定使得编码生成的dna序列高效地满足了序列限制的条件。由于使用的是10个双碱基码字,没有任何码字的两个碱基相同,因此无论码字怎么排列,最多相邻的两个码字的相邻碱基相同,不会出现连续三个碱基相同情况。除了at和ta,其它八个码字都是gc和at各一个,因此gc含量会比较均匀。将at和ta定义为编码8和9,是因为13位的二进制数据最大是8191,因此绝不可能出现8888,8899,8998,9999等只有8和9组成的四位数,也就是说任意相邻的四个dna码字,至少有一个码字含有g或c,这样在任何情况下,编码序列中都不会有超过8个碱基的序列全部是at的情况。由于只含gc的双碱基码字全部排除,所以任何情况下都不可能有超过两个碱基的gc序列。
46.极端情况下从二进制到十进制的转换结果中,可能会出现类似于1888199918981889...这样的数字串,这种情况下会导致gc比例过低,只有12.5%。这种情况下采用码字修正,将gc和cg分别作为at和ta的替补码字,仅仅用于在任何连续的两个或三个8或9时,选择性的使用gc代替at,或者cg代替ta,编码其中一个或两个数字,即可彻底解决gc含量过低的问题。
47.以上通过简单的字典定义和规定,充分地保障了二进制数据转换到dna碱基后的任何的可能,即使最极端的情况下也绝对保证序列的单碱基重复不超过2个,gc含量会稳定在50%上下的狭窄范围内,这样彻底解决了转换后的dna序列限制问题。
48.信息分段:将待存储二进制信息分段,分段长度为13比特的倍数,序列长度的确定参考商业公司dna合成服务报价,一般情况下为150碱基或者200碱基长度,后者仅仅比前者价格高不到5%,但是却增加了33%的序列长度,因此性价比会更高。200碱基长度中,两端引物一般至少要使用40碱基(20碱基*2),8碱基序列纠错码,24碱基地址索引,剩下128碱基,正好是8碱基*16,可以编码208比特信息(13bit*16),这种设置相对比较合理。因此本实施例中将信息按照208比特分段,合计26个字节。
49.rs纠错码是非常成熟的纠错码,它基于本原多项式通过特定代数运算来纠正传输信息过程的突发错误。由于其编码解码原理算法和本发明无关,并且是教科书级的内容,这里就不做陈述,只是使用它。其基本纠错能力表述为,当发送信息字数为k个时,添加m个冗余字,将这k+m个字的信息发出后,接收方能够纠正这k+m个字里的最多任意m/2个字的错误。本发明使用rs纠错码生成冗余信息来供读取数据时的纠错,根据所需要的冗余比例冗余(如15%),产生冗余分段(如使用850个信息分段产生150个冗余分段,或8500个产生1500个等)。冗余分段是按照固定长度比特生成,其内容由所有信息分段的对应字节内容根据rs(里德所罗门)纠错码生成规则生成,如:使用所有信息分段的第一个字节根据rs纠错码规则生成150个字节,组成150个冗余分段各自的第一个字节,然后依次对第2~26个字节进行相同处理,生成150个冗余分段。冗余分段生成结束后,冗余合计占分段总数的15%,编码分
段占85%。每1000个(或者10000个等其它数量)分段中相同位置的所有字节,比如第一位的1000个字节,构成一个冗余纠错基本单元。
50.进制转换:按照每13比特转换为一个四位十进制数,将所有的分段208比特二进制数据转换成为64位的十进制数字串。13比特的二进制数最小为0,最大为8191(2
13-1),四位十进制数的空位用0补齐,如:1补齐为0001,28补齐为0028,487补齐为0487。最终结果将208位二进制数串转换为64位10进制字串。
51.序列编码:根据字典使用双碱基dna码字替换二进制序列数字,将64位十进制字串转换为128位碱基dna序列。根据字典和编码规则,能够保证在任何情况下这些序列都符合序列限制的要求,gc比例均衡且没有超过2个碱基的单碱基重复。
52.添加地址索引:根据字典,使用双碱基dna码字编码阿拉伯数字,生成一个24碱基长度,编码12位的十进制碱基数字串,将其添加在128碱基序列前面作为该碱基序列的地址索引,每一条128碱基dna序列使用不同的12位十进制碱基数字串来记录这个序列的精确位置,生成152碱基长度序列。
53.12位十进制数字串共有10
13
个,按照每条序列26个字节,15%冗余计算,理论上单组dna可以存储220tb(26*85%*10
13
byte)。一般来说tb级容量够用,再加上可以使用不同的引物组合,所以可以考虑使用11个码字编码数字,另外一个码字当作校验码,比如前面十一个数字之和,模10的余数是多少,这个校验位数字就是多少,可以检测到地址索引的单个数字错误,从而排除错误定位的可能。单组dna容量上限降低到22tb,已经足够应用。
54.地址索引的数字排序,从0开始到10
11-1,加上一个校验位检错,转换成dna序列也符合序列限制的要求。全重复数字转换后,也仅构成两个碱基的重复,dna序列也能够符合序列限制的正常要求。
55.添加纠错码:在上一步生成的152碱基长度序列后添加8个碱基长度的校验位,得到160碱基长度序列,校验位的碱基按照汉明码和rs纠错码的各自规则添加即可。
56.根据汉明码规则,使用8个校验位可以校验最多247个数据位(2
8-1-8),并且可以纠正255个数据(8+247)中的任何单个数据错误。而7个校验位,可以校验最多120个数据位(2
7-1-7)。而前面步骤生成的序列长度为152个碱基,所以必须使用8个碱基的校验位。
57.添加汉明码校验位时,忽略前面所有的信息编码内容和地址索引等,将152个碱基看成一串acgt的四进制数字,按照统一标准将四种碱基分别一一对应0/1/2/3。并且根据统一标准将其依次填入8碱基校验位所对应的247个数据位中,余下的空位丢弃,将152个碱基数字依次填入后,根据汉明码规则,8个校验位分别放在2n(n从0到7)的位置上,每个校验位所应其校验的所有数据位的数据之和模4的余数(0/1/2/3),校验位填入按照统一标准对应的碱基,获得8个校验位碱基后,依次将其直接添加到152碱基后面即可,这样序列变成了160碱基。这个汉明码是基于四进制的,因此是四进制而不是二进制汉明码。如采用二进制,则将碱基的两个比特分开,所有碱基的前一个比特构成一个汉明码,后一个比特构成另一个汉明码即可。
58.需要说明,8碱基序列按照汉明码里面的位置信息分别插入到相应的实际位置,效果也完全相同,只要统一标准明确哪个碱基在虚拟的汉明序列里位置即可,对结果并无影响。这8个碱基的序列各种可能性都有,因此也可能出现都是gc或者都是at,或者都是单个碱基的重复,由于仅有8个碱基长度,所以实质上可以接受,正常情况下,生物的dna序列里8
个碱基的任何序列都会经常出现,相对比较短,不会显著影响合成和测序。
59.添加rs纠错码(此处为单条序列纠错码)时,按照每四个碱基对应8比特为一个字符的原则添加两个字符,共8个碱基来纠正所有40个字符(包括纠错码的两个字符)的任何单个字符(连续4个碱基)错误。
60.添加两端引物:在160长度碱基长度首尾两端各添加20碱基长度的引物,得到200碱基长度dna序列。
61.针对所有序列的引物都相同,也可以根据编码信息的实际内容使用不同的引物,以便随机读取个别文件内容。如果引物不同,就需要在前面步骤避免同一条序列上写入两个文件的内容。一般情况下,目前的数字化文件结尾都有专门的文件结束符号,若单个文件长度最后的若干个字节不够一条全长序列,剩下的空余位直接全部填0即可。
62.合成dna并测序:把前述步骤所获得的所有dna序列以阵列芯片的方法进行合成。
63.dna合成获得寡核苷酸池干粉或者溶液,其总量较少,10万条片段的质量大约1微克。至此完成数据到dna分子的写入,可以直接长期保存,也可以通过pcr扩增出产物后保存pcr产物。微克级别的寡核苷酸混合物可以进行至少大约100次左右的pcr,每次pcr理论上可以扩增出所有的片段,以指数级别增加所有片段的数量并将其变成双链dna,然后按照商业公司的标准,取部分pcr产物交给商业公司测序,即可获得这些片段的dna序列。
64.数据解码:如果获得的理想测序结果完整且没有任何错误,即可逆向转换恢复全部数据。但实际测序结果和原始设计的dna序列会有差距,主要表现在两个方面,一是实际测序结果和设计的序列相比会有大约0.5%到1%的碱基错误,这些错误主要是由于合成寡核苷酸时的随机错误造成。二是不同dna序列片段合成后不均匀,测序结果中会显示有的序列数量多,有的数量少甚至缺失。所造成的结果就是在一定测序深度下会有一定百分比的序列丢失,而增加测序深度又会增加成本,所以解码数据需要首先解决上述两个问题后,才能获得全部数据。
65.假设片段碱基错误率是1%,以10倍深度测序获得结果。10倍深度意味着假如一组dna有1万个片段,那么通过随机测序获得10万个分子拷贝的序列,平均下来每个设计合成的dna序列都被测了10个分子拷贝。然而实际上会有一些序列被测了几十个分子拷贝,而另外一些只测出1个分子拷贝,还有的根本就没有被测出。这是由于前面所述的合成时,目前的技术还无法保证成千上万个序列以完全一致的平均规模合成所致。目前现有的编码方法研究中,测序都需要至少百倍到数百倍甚至上千倍的深度来确保完全恢复数据,但喷泉码和混合进制方法由于结合了冗余和纠错的方法,因此获得百倍深度测序结果后,尝试从测序结果中用电脑程序模拟,随机抓取低倍深度的测序序列,来测试最低需要多少倍深度仍能完美解码,结果是分别仅需要10倍和5倍深度即可。喷泉码设计了序列rs纠错码,可以纠正每条序列的最多1个碱基错误,并利用其编码原理生成了7%的冗余序列,用于解决序列丢失的问题。而混合进制方法则没有单条序列的纠错码,使用了15%的rs纠错码冗余,同时解决碱基错误和序列丢失的问题。
66.本发明同时使用了序列纠错码和多个序列间的rs码冗余两个维度纠错码,分别用于单条序列纠错和多条序列之间的冗余纠错,因此会比上面两种编码方法只使用一种的效果会更好一些。其中汉明码根据规则可以纠正序列中单个碱基的错误,但不能纠正两个碱基的错误。由于任意汉明码码字之间的汉明距离至少是3,因此也能够检测到两个碱基的错
误。汉明码只能检测到错误,并不能直接区分是一个错误还是两个错误。如果错误数量为1则会被改正,如果错误数量为2则会强行纠错,导致出现三个错误。在10倍深度测序结果里,根据经验可知设计序列中的至少95%都可以被测出至少一个分子拷贝,喷泉码的文献中,使用10倍高质量测序结果恢复了全部数据,根据其解码原理的要求,可以反推其至少应该有95%的序列被测出,并且经过序列rs纠错码纠错后,所有的错误都被纠正,才能根据其喷泉码原理使用这95%序列完美恢复出所有存入的信息。
67.本发明在同样使用能够纠正单个错误的纠错码时,也能够获得95%的完全正确序列。即使退一步,只能有90%的序列测出了至少2个分子拷贝并且经过纠错后完全正确,也足够使用。按全部序列中至少90%是完全正确,5%存在可能错误的情况分析rs码纠错,根据前述碱基错误率1%的设定,5%中的错误碱基占所有碱基总数的0.05%。考虑最糟糕的可能,单个碱基错误导致8碱基一组编码的13位比特数据错误,这又会最多造成三个字节(8比特*3)的错误,比如13个比特被分到了三个字节里面。所以这最多会造成1.2%的字节错误(0.05%*8碱基*3字节),因此这5%里面会有最多总数1.2%的字节错误,正确的最少有3.8%,加上前面的90%,共有93.8%的正确序列的字节。考察之前的设定,根据地址索引可以将95%的序列进行精确定位,因此也就能明确知道剩下的5%缺失片段的位置。
68.使用rs纠错码恢复全部数据,根据前面步骤设计的rs码纠错,一个码块是1000个所有序列的相同位置8比特字节,每个位置共计1000个,缺失了5%就是50个,填上任意数据,并且当作它们都是错误数据对待。冗余设计的是15%,也就是150个冗余字节,根据rs纠错码规则可知,能够纠正的错误字节数量是75个,而目前的错误字节最多只有62个(1000-938),所以完全有能力进行全部纠错,同理纠错第2个码块直到第26个码块。如果为了应付序列可能的随机错误局部密集,只需将rs纠错码的一个单位扩大即可,比如每10000个序列的第一个字节,混合进制方法的一个rs码单位超过6千多条序列,使用了15%的冗余,最低测序深度5倍即可解码。同时也可以看出,即使前面所述90%的全部正确序列里,仍然有很低比例的错误没有被纠正,也会在rs码纠错的过程中被完全纠正,只要错误比例在允许的范围内。和混合进制编码方法相比,本发明增加了序列纠错汉明码,所以理论上至少不会比其纠错能力差。
69.进一步说明本发明的序列纠错码纠错,也可以使用rs纠错码,本发明将汉明码从二进制拓展到四进制,效果也基本一样。如果一条序列测出了两个分子拷贝,各自带有2个错误,经过比对,如果错误位置不同,这两个分子会有4个位置不同。假设甲分子这四个位置的碱基为abcd,乙为abcd(为了看起来更清楚,这里用字母代替碱基)。那么可以得出结论,甲的两个错误碱基只能是下面六种可能性之一;ab,ac,ad,bc,bd,cd。参考乙的相同位置,如果甲的碱基错误了,这个位置正确的碱基应该是乙的对应位置碱基。假设a错误,a正确,那么考察甲分子,将其改为abcd后,序列中应该会只剩下一个错误,且必定是bcd三个中的一个。利用汉明码纠错就应该能够将其中之一纠正为小写的bcd之一,否则a就不是错误碱基,继续逐个考察bcd。结合汉明码的特点,两个汉明码的汉明距离至少为3,使用这种推理就可以确定无疑地纠正两个分子拷贝的各自两个错误,不论这两个错误在什么位置。如果甲有一个错误乙有两个,或者甲没有错误乙有两个,或者甲乙合计有四个错误,都可以推理出正确的碱基序列,这里就不一一逐个分析。尽管汉明码只能纠正一个错误,但是在两个不同分子拷贝的参考序列加持下,就可以完全纠正各自的两个错误。而根据经验估计,10倍测
序深度会有90%的序列都会有至少两个分子拷贝被测出,而按照1%错误率,160个碱基长度序列的错误数量实际平均只有1.6个。
70.如果不使用序列纠错码,哪怕两个分子拷贝序列中,一个拷贝错了一个碱基,另外一个分子拷贝完全正确,也无法判断出正确序列到底是什么,只能通过深度测序测出更多拷贝,通过多数投票原则确认正确序列。且多数一致的碱基存在错误也无法获知,但序列纠错码可一票否决,即使测出100个分子,序列完全相同,纠错码显示有错,那必然有错。对于序列测序结果中有碱基缺失的情况,根据统计数据,碱基缺失大约是碱基错误的十分之一左右。如果这个序列有多个分子被测出,那么可以通过互相比较确定出缺失位置,如果只有一个分子,并且缺失碱基,可以先置之不理,当作序列缺失对待即可。简单计算可得知,只有单分子被测出且碱基缺失的,在总体测序结果中的比例远低于百分之一。
71.调整比特模块:本发明采用2个碱基码字编码十个阿拉伯数字,再使用13比特换为4位十进制数,然后再将十进制数字串按照字典转变为碱基序列。上述转换方式还进行调整,比如将3比特二进制数转换为一位十进制数字,6比特二进制数转换为两位十进制数字等。这种转换和三进制编码的本质差别在于,若干位二进制比特能表示的最大数小于若干位的十进制能表示的最大数,3比特的最大数7(111)小于9,6比特的最大数63(111111)小于99,13比特的最大数8191(1111111111111)小于9999等,因此初始二进制数据不需要经过哈夫曼压缩减少字符数。否则,本发明可使用10比特二进制(最大数1023,2
10-1)转换为三位十进制(最大999),通过哈夫曼压缩减去24个字符即可,信息密度可以达到1.67(10比特/6碱基),而13比特转换为4位十进制数的信息密度为1.625,都高于三进制的1.6。一般来说,原始数据都需要无损压缩减少体积,因为dna存储的成本太高,而哈夫曼方法的压缩效果远不如其它压缩算法,因此三进制实际上要在其它压缩算法压缩后,再次进行哈夫曼压缩来满足其编码的要求,往往达不到压缩的效果,仅仅为了减少字符。更加需要说明的一点是,哈夫曼压缩减少字符后,无法再进行rs码纠错冗余,因为冗余会产生新的已经被减去不能使用的字符。
72.将3比特二进制数转换为1位十进制数字时,其存储密度和混合进制的编码存储密度效果完全相同,但序列转换方法明显不同。三比特只有八种,用0到7八个数字即可,双碱基则使用ac,ca,ag,ga,gt,tg,ct,tc这八个。这八个码字的使用可以保证其编码的任何序列,gc含量都固定为50%,并且绝对不会出现连续超过两个碱基的重复,而混合进制显然做不到这一点,其合法码字就有4个三碱基重复的码字。
73.序列限制:本发明字典中有8个码字自身就是gc含量50%,另外两个码字at和ta又分别使用gc和cg备份,用于调整可能出现的at或ta连续码字。如当相邻的两个阿拉伯数字是8或者9时,使用gc或cg中的一个码字来编码8或者9,来保证序列gc含量平衡。这样使得在任何情况下,生成的dna序列中,任意超过10碱基长度的序列,其gc含量都绝对保证在40%-60%,整体上稳定在50%左右,也不会出现超过两个碱基的单碱基重复。这不同于三进制和混合进制方法,例如在三进制方法中,gcgcgc......或atatat......都是合法的序列且无法调整,显然gc含量太高或太低。而混合进制方法中,gggngcgnggcn.....(n表示a或t)或aatnaaanattn.......(n表示g或c)也是合法序列且无法调整,这样的gc含量也会达到75%,或者低到25%。
74.容量和质量:存储数据的单组dna按照平均每个序列含有1000个分子拷贝计算,使
用200碱基长度的寡核苷酸,经过pcr变成双链,存储1tb数据的dna的干粉质量大约10mg,使用毫升体积的容器即可以保存,采用尺寸略小于手掌(15cm*10cm*3cm)的常规分子生物学96孔细菌培养板即可保存96组dna。也有研究人员建议使用滤纸,将dna溶解后,液体滴到滤纸上干燥后去除水分,这样就可以大大缩小保存体积。还可以将dna封装在二氧化硅(如玻璃)内部,隔绝空气和水分长期保存。
75.如果使用引物组合增加单组dna容量,就很难做到将tb级的数据保存在10mgdna中。如前所述,由于不同序列的分子合成的数量不平均,假设pcr扩增产物,模板需要平均每个序列100个分子才能保证有足够的覆盖。那么使用本发明方法,平均每个序列含有1000个分子拷贝的一组dna保存数据,每次取十分之一当作模板就可以使用10次。但是如果使用1000对引物组合(其它方法单组dna容量低于1gb)达到tb级别容量,引物组合的多重pcr,即使按照多至50对引物一个pcr反应,那么使用10次后,也只能扩增出500对引物的数据就耗尽了dna,还有一半的数据就无法得到,因此只有增加dna的总量才行。这个问题的原因是,本发明的一对引物的pcr,可以将当作模板的所有分子都扩增出来,但使用引物组合的pcr,取出的模板dna分子中,绝大多数dna分子由于引物序列不同并不能被扩增,因此白白浪费,所以只能增加单组dna的量,结果会导致dna的质量密度大大降低。
76.冗余比例调整:本发明实施时将冗余比例设置在15%只是一个低倍深度测序下的理论参考值参考值,实际上可以根据实际情况调整。目前相对于合成来说,测序的成本要低很多,因此可以参考性价比来设置冗余比例。经过足够深度测序,可以获得99%以上的正确序列,这种情况下所需的冗余比例就可以降到很低。
77.再例如,可以考察将寡核苷酸池通过pcr扩增后,取少量pcr产物当作模板再次进行pcr,这样重复十次到几十次,最终测序检测序列会有多少比例彻底丢失,保证在这个情况下还可以恢复全部数据来设置冗余比例。还可以考察在各种条件下的dna半衰期情况,研究时间对dna序列损失的影响,这样就可以调整冗余比例,为数据安全保存数百年到万年之久留有充分的余地。
78.性能比较:三进制,混合进制,喷泉码,阴阳码和本发明的信息编码区单碱基比特密度依次为1.60,1.50,2.00,2.00,1.63。如果考虑到包括引物在内全长寡核苷酸,并将各自的序列冗余计算在内,实际密度则分别为0.29,0.80,1.17,1.02,0.88,可以看出本发明仅仅低于喷泉码和阴阳码(以上数据来源根据相关文章中的数据实际计算获得)。其中三进制由于采用重叠4倍覆盖,降低4倍后再考虑引物在内,所以实际密度只有0.29;阴阳码的缺点是无法百分百恢复数据,实际上存储了仅约0.5m的数据,并且测序深度也足够,却仍然只能恢复99.9%的数据,不可避免地会丢失少量数据。喷泉码和混合进制分别实际存储了2m和200m的数据,却一个比特都不差地恢复了全部数据,分别模拟最低10和5倍测序深度也可全部恢复。本发明方法根据目前的寡核苷酸池合成的实际测试的质量数据,理论上推理出在设置rs码15%冗余序列纠错的前提下,使用约10倍深度的测序数据,即可百分百完全恢复数据。
79.喷泉码高达1.17的密度源于其编码区的单碱基2比特的极限转换值,但这种依靠喷泉码的方法,需要依赖于计算机的强大算力。相关文章中指出,在rs纠错得到正确序列后,根据喷泉码规则还原数据时,使用普通的苹果电脑需要9分钟恢复大约2m的数据,这过于复杂;而其它方法纠错后就可简单还原数据,作为参考,目前的数据压缩的算法采用普通
苹果或英特尔的计算机解码压缩数据的速度一般可以在每秒几百mb数据上下范围。因此喷泉码存储高密度的代价是电脑计算力,尽管看起来的确是代表目前世界的最高水平,但应用前景并不被特别看好,因为难以想象tb级别的数据恢复会需要多少时间。微软公司资助华盛顿大学的研究人员提出了混合进制的6比特转四碱基的编码方法,而本发明的密度比其高约10%。
80.本发明的单组dna最大容量上限,高于其它使用地址索引的方法至少千倍,且可以通过增加地址索引的碱基序列长度增加容量,每增加两个碱基即可提高10倍,足以满足实际应用的任何容量要求。尽管多种编码方法作者文章中都给出了dna存储的极高质量密度,比如喷泉码是21eb(eb:10
18
b)每克dna,阴阳码给出高十倍(in vitro)或百倍(in vivo)的密度值,实际上并无实际应用价值,因为这些编码方法的单组dna容量上限,在不使用引物组合的前提下,均不超过1gb(109b),即使使用引物组合,也就在1tb到几十tb之间,且dna存储要考虑到实际应用的需要,一般毫克级一组dna保存比较合理。
81.所以理论上,假如将超过eb的数据按照其它编码方式保存在1克dna并且这些dna混在一起,然后给出全部的正确dna序列,这些编码方法也无法解码,这源于其方法的单组dna容量受到限制。但是本发明适当调整,将地址索引增加长度,如到40碱基长度来编码10
21
个索引地址,就可解码zb级数据(1zb=1000eb),相应信息编码长度减少,也只是按比例降低了大约15%的编码效率而已。其它方法的地址索引调整难度极大,如果强行调整,关于索引的数据也会是海量,并且需要存储在电脑系统等目前的各种电子存储设备中,以供解码时参考。那么用高额成本将数据存储在dna中就失去意义,还不如将这些数据直接存在电子设备中。
82.本发明的地址索引的码字和编码区完全相同,哪怕地址索引有重复数字,编码后得到的dna序列按照喷泉码和混合进制的序列筛选标准评估也都符合要求,所以可以直接按照十进制的自然数列来记录地址索引,但考虑到实际可能带来的问题,可以适当改进,采用一个和10互质的数p作为基本数字,按照逐个增加p而不是1来计数,保留和索引码字相同长度的位数,若产生更高进位数直接丢弃超出长度的高位数,这样作为计数序列使用。如考虑两位数计数,自然数序列是01,02,03,04.....99。改用这种方法,采用质数91为计数单位表示相邻两个数的差,其序列位91,82,73,64,55,46,37......。这样做的原因是,假如自然序列的前面数字,编码后许多数位都是0,这样编码后得到的双碱基重复序列可能相比较其它序列更容易出问题,因此可能会造成这一组rs纠错码块的数据恢复困难。但是采用这种以一个和10互质的大数为计数单位排列时,会将那些双碱基重复序列,也就是索引是单个重复数字的索引分散开到不同的rs纠错码块,从而尽可能分散其影响。
83.改进其它编码方法:本发明索引和数据编码采用同一套规则,简洁高效。如果将本发明地址索引和思路用于改进其它方法,也将取得非常好的效果。
84.喷泉码使用16个碱基的索引,只能检索大约1.6*107个地址(根据其文章中数据,最大容量500m和每条序列保存32字节计算出)。假如采用本发明地址索引的基于10进制转换方法,16碱基可以保存8位十进制数字,因此检索地址总数量为1*109个,比它的方法高约60倍,同时即使采用自然数序列计数,生成的dna序列,也完全符合喷泉码评估dna序列的标准。喷泉码使用了24碱基长度引物,如果像阴阳码和混合进制编码方法,使用20碱基长度引物,两条引物一共可以节约8碱基,用于地址索引,那将会产生12位十进制的索引,索引数量
将达到1*10
13
个,单组dna容量理论上限会达到约300tb,考虑增加7%冗余序列后平均单条序列实际编码约240比特。
85.混合进制使用14碱基地址索引,假如使用本发明的方法,会产生7位十进制的索引,索引数量将达到1*108个,同样明显高于其自身的索引地址产生方法。再进一步使用更长的dna片段长度,采用200碱基而不是150碱基,前者比后者增加50%长度而商业报价仅仅高不到5%。使用140碱基编码序列编码210比特,20碱基地址生成1*10
11
个索引,依然使用15%的rs纠错码,单组dna容量理论上限会达到约2tb,平均单条序列实际编码约183比特。
86.三进制本身采用了4倍覆盖片段重叠的方法,假如采用本方法的地址索引和编码思路,不采用8比特转换为5碱基,而采用11比特转换为7碱基。这种11到7的转换可以避免哈夫曼压缩减少字符的步骤(从256个字符减少到243个字符),因为2
11
=2048,而37=2187,2048小于2187,因此11比特转换7碱基就可以直接转换。规避了哈夫曼转换的步骤后,就能够采用rs码冗余,按照15%的序列冗余,10倍左右测序深度,即可100%恢复所有数据,而原方法在4倍覆盖冗余300%和数百倍测序深度的前提下,依然不能恢复所有数据。这样改进后,使用126碱基(7碱基*18)保存198比特(11比特*18),26碱基地址索引,8碱基纠错码,加上15%序列冗余,单组dna容量理论上限会达到约2pb,平均单条序列实际编码约172比特。
87.而阴阳码仅仅使用了16比特的地址索引,因此最多能够生成大约6.5万个地址,又将两段二进制融合到一段dna序列,再加上了20%冗余(每4段二进制增加1段冗余),综合计算,它的单条序列实际编码大约205比特,单组dna的理论最大容量仅大约800kb(128bits*6.5万*80%),显然远远低于其它所有方法,根本达不到实用的要求。其独特的二进制数据到碱基序列的转换模式,使得本发明的地址索引方法无法直接用于改进它。基于其创造性地使用两套编码系统将两段二进制信息融合到一段dna序列,并且过程可逆的基础上,仍然可以调整改造其编码流程来使其达到实用性高的目的。首先需要摒弃它编码方法的1536种不同策略,将其缩小为64种。具体就是去除编码过程中,根据前一个碱基的不同而有24种(4!,4的阶乘)不同的组合,因此将策略缩小为64种(1536/24)。摒弃这点的原因是,假如某个碱基出现错误,将会在解码过程中,造成这个碱基后面的这个碱基也会解码错误,从而增加后续纠错步骤负担。摒弃后则把单个碱基的错误固定在这个碱基本身,而不影响其后碱基的解码。其次将纠错码调整为使用整体rs纠错码纠错,不再使用单条序列的纠错码,因为这样做的整体纠错效率更高。第三是增加地址索引的比特数到30比特,使用128比特存储数据,合计158比特,整体两段158比特转换成158碱基序列,再加上2个碱基校验是否有单碱基错误。根据这个思路改进后,能够确保阴阳码在低倍深度(比如10倍)即可恢复100%数据,单组dna的容量上限可达到20gb,平均单条序列实际编码大约218比特,这三个数据均显著高于改造前的99.9%,800kb和205比特。
88.阴阳码改进后的具体方法步骤为,首先将待存储的二进制信息按照128比特分段和本发明一样,先按照15%比例生成128比特相同长度的rs码冗余分段。其次给所有的分段,包括冗余分段,按照先后次序在前面添加30比特的地址索引,得到158比特的分段。第三选择64种不同组合的阴阳码的一种,进行编码,转换成158碱基的dna序列,由于将两段二进制融合成了一段dna序列,因此分段的总数量将会减半。最后将158碱基,按照相邻碱基每两个一组,共79组,转换成16进制(4*4)数字,79个数字后相加之和,模16后得到一个余数,再用两个碱基来记录这个余数,就构成了一个校验位,可以检测任何单碱基的错误,或者80组
双碱基中的任何一组双碱基的任何错误,以及大部分任意两个碱基或多个碱基的错误。至此编码完成,合成dna后,测序逆向解码即可恢复全部数据。其中30比特的地址索引可以直接从0开始按照自然数顺序排列即可,连续的0和1,对后续通过阴阳码来转换后碱基序列的影响,基本可以完全忽略不计。
89.以上几种目前dna存储领域最好的几个编码方法,经过本发明的方法或者思路改进后,总体性能大大改进,在同等条件下,即使用200碱基长度的寡核苷酸,20碱基长度的引物条件下,改造后的编码方法和本发明编码方法(按照尽量公平原则,本发明使用24碱基地址索引和136碱基编码信息,整体rs码序列冗余纠错),将全长200碱基作为考量基础,平均的单碱基实际存储信息密度(比特/碱基),平均每条序列实际存储信息比特数和单组dna的理论容量上限分别为如下数据,改造后这些方法全部都能100%恢复所有数据:
90.喷泉码,密度为1.17,每条序列实际存储约240比特,容量上限为300tb;
91.阴阳码,密度为1.09,每条序列实际存储约218比特,容量上限为20gb;
92.三进制,密度为0.86,每条序列实际存储约172比特,容量上限为2pb;
93.混合进制,密度为0.91,每条序列实际存储约183比特,容量上限为2tb;
94.本发明,密度为0.96,每条序列实际存储约181比特,容量上限为220tb;
95.其中容量单位换算关系约为,1pb=1000tb,1tb=1000gb。
96.从以上数据中可以看出,其它方法按照本发明的方法或思路改进后,考虑性价比和实用性上,优秀程度排序大体上依次应该是:喷泉码、本发明、混合进制、三进制和阴阳码。阴阳码排在最后,是由于其单组dna的容量上限仅仅为20gb,达不到tb级别的实际要求。喷泉吗的最大弱点是对计算力的依赖性太强,况且随着存储数据量的增加,所需计算力很可能不是线性级增加,而是指数级增加,这就对它的应用前景披上了极其不利的阴影。
97.本发明通过阐述过程和原理,结合实际,包括寡核苷酸合成真实数据,纠错算法本身的能力,也讨论分析了一些细节,清楚明确地证明了本发明的可行性。
98.目前的各种dna编码方法,其单组dna的容量受到很大限制,本发明的一个重点就是解除了这个限制,且编码规则简洁,高效地解决了单组dna容量和单碱基信息密度之间的矛盾,综合性能优于目前其它的dna存储编码方法。
99.编码汉字文本:作为本发明编码方法的应用扩展,本发明编码方法可以直接编码汉字文本,首先使用0000到8191(2
13
)这些四位数编码汉字。国标gb2312规定的简体汉字共有6763个,加上阿拉伯数字,各种标点符号和大小写英文字符等不到100个,合计约6860个左右。而其它的不常用汉字或者繁体字,采用字头字符+普通字符的双字符来编码,从其它剩余的四位数中选20个当作字头字符,和6763个汉字的字符,共有超过13万多个双字符组合,足够表示所有不常用汉字和稀有汉字。另外还会剩下超过1千多个四位数留作备用,比如选择部分来表示拉丁字母或其它语言的普通基础字符。对于使用频率非常高的汉字或者字符如“的”、“一”、“是”、逗号、句号等,都可以使用多个四位数编码来适当地增加序列的多样性。
100.当代很多汉字文本都会有夹杂英语单词的情况,而使用一个四位数表示一个字母又会比较浪费,因此还可以使用若干个四位数当特殊符号,表示其后的每个四位数表示两个字母,例如使用7240当特殊符号表示其后的四位数编码两个字母,那么72405624即表示56和24分别表示一个字母;7241表示其后的两个四位数表示四个字母,724156241359即表
示56,24,13,59四个字母,7242表示其后的三个四位数表示六个字母,以次类推,只要另外再将字符和空格分别用2位数定义即可,当单词字母为奇数个时,用空格补在最后。这样汉语文本中的外语单词也就会比较紧凑。总之剩余的一千多个四位数可以灵活定义,以应付各种情况使用。
101.对汉字文本也采用同样编码解码流程,先将文本按照16个汉字(四位数)一段,先进行rs码冗余一定比例的分段,然后添加地址索引后,按照字典将数字转换为碱基序列即可。
102.基于十进制的dna存储编码设备与可读存储介质
103.本发明提供基于十进制的dna存储编码设备,包括:
104.存储器,用于存储计算机程序;
105.处理器,用于执行所述计算机程序时实现如权利要求1-4任一项所述基于十进制的dna存储编码方法的步骤。
106.上文所描述的dna存储编码方法的步骤都可由该设备的结构实现。
107.相应于前述方法实施例,本发明还提供一种计算机可读存储介质,计算机可读存储介质上存储有计算机程序,计算机程序被处理器执行时实现如前述基于十进制的dna存储编码方法的步骤。
108.以上示意性的对本发明及其实施方式进行了描述,该描述没有限制性,附图中所示的也只是本发明的实施方式之一,实际的结构并不局限于此。所以,如果本领域的普通技术人员受其启示,在不脱离本发明创造宗旨的情况下,不经创造性的设计出与该技术方案相似的结构方式及实施例,均应属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1