一种基于定向基因组测序获取全基因组SNP位点的方法
本发明涉及遗传学领域,具体说是基于定向基因组测序技术获取全基因组snp标记位点的方法。
背景技术:
1、单核苷酸多态性(single nucleotide polymorphisms,snp)是目前最重要的分子遗传标记,因其在基因组中大量广泛分布,具有极高的检测丰度,且遗传稳定性较高,在分子遗传学中占据了重要地位。在人类和动植物遗传育种领域中,snp被广泛用于复杂性状遗传机制的解析、基因组选择等研究方向。
2、不同的研究内容对遗传标记的数量需求有所不同,在遗传育种应用方面提高标记数目仍然是研究的重点之一。例如,基因组选择(genomic selection,gs)利用覆盖整个基因组的高密度snp来构建亲缘关系矩阵,从而估计候选个体的基因组育种值并进行选种,因此需要通过高密度的遗传标记捕获到更多的加性遗传变异;而在全基因组关联分析中,想要更准确地鉴定到目标表型真正的致因突变,也需要提高标记密度。
3、当前主要的全基因组高通量分型技术有snp芯片和基因组测序两种。snp芯片的优势在于操作简单、分型速度快、准确性高,对分型流程的环境设施要求较低,可以在生产一线直接应用,因此芯片分型成为早期全基因组分型的主流方法。其不足之处在于,芯片设计完成后,能鉴定的snp数据集是固定的,难以满足所有类型的研究需求,后期不易拓展升级,并且当面对多个育种群体时,部分snp在有些群体中已经固定,因此同一款芯片难以覆盖所有群体的遗传特点,导致一些群体遗传信息损失。
4、测序成本不断降低使大规模的测序在动植物育种领域得到广泛使用。而定向基因组测序,意即将感兴趣的基因组区域通过探针杂交、酶切富集、多重pcr扩增等方法进行富集后进行测序的研究策略,根据不同的应用,利用较少的数据量就能得到超高的灵敏度和准确度,实现变异位点的快速筛选。目前定向基因组测序的方法有很多,例如简化基因组测序(genotyping by sequencing,gbs)、靶向基因组测序、多重pcr测序等。这些定向测序在不同程度上存在的共性问题是,基因组不同区域的富集效率可能存在较大差异。利用现有分析手段对该问题最常见的解决办法是,将低富集区域的测序数据进行删除,只保留高富集区域的测序数据用于基因组突变鉴定,这就直接损失了很多本应该富集到的基因组目标区段。为了在测序数据中能够获得更高比例的高富集区域,一般通过提升基因组整体测序深度来保证,然而这大幅提升了测序成本。
5、以gbs为例,这类方法通过富集并测序基因组中很小比例的片段得到分布全基因组的snp标记,同时实验流程简便且成本较低。tassel软件是基于简化基因组开发的程序包,专门用于gbs数据的snp检测,但是对于双端测序而言,tassel无法直接对双端reads识别,需要对每端的reads单独处理,处理后,要对两端的reads进行合并。并且tassel在发现变异位点的过程中存在将插入缺失变异(indel)误判成snp的可能,因此在用传统的tassel程序对gbs数据进行变异位点发现时存在假阳性。此外,传统gbs分析流程,对于每个位点靶向测序的测序深度存在最低要求(一般均要求10×/位点以上),对于部分未达到测序深度要求的位点,则无法准确判定其杂合子/纯合子的状态,如果删除该位点则损失了标记密度,如果保留该位点则很可能降低分型的准确性,准确性较低的分型结果会进一步影响到填充准确性,不利于育种实践。
技术实现思路
1、针对现有技术中存在的缺陷,本发明的目的在于提供一种基于定向基因组测序获取全基因组snp位点的方法。利用本方法产出的定向基因组测序数据,目的在于提供一种新的定向基因组测序基因分型方法,在维持低成本的同时,能够获得更高密度的snp标记,并提高分型的准确率。
2、为达到以上目的,本发明采取的技术方案是:
3、一种基于定向基因组测序获取全基因组snp位点的方法,其特征在于,包括如下步骤:
4、步骤1,提取育种群体中待检测个体样本的基因组dna,对上述基因组dna进行定向基因组测序得到定向基因组原始测序数据;
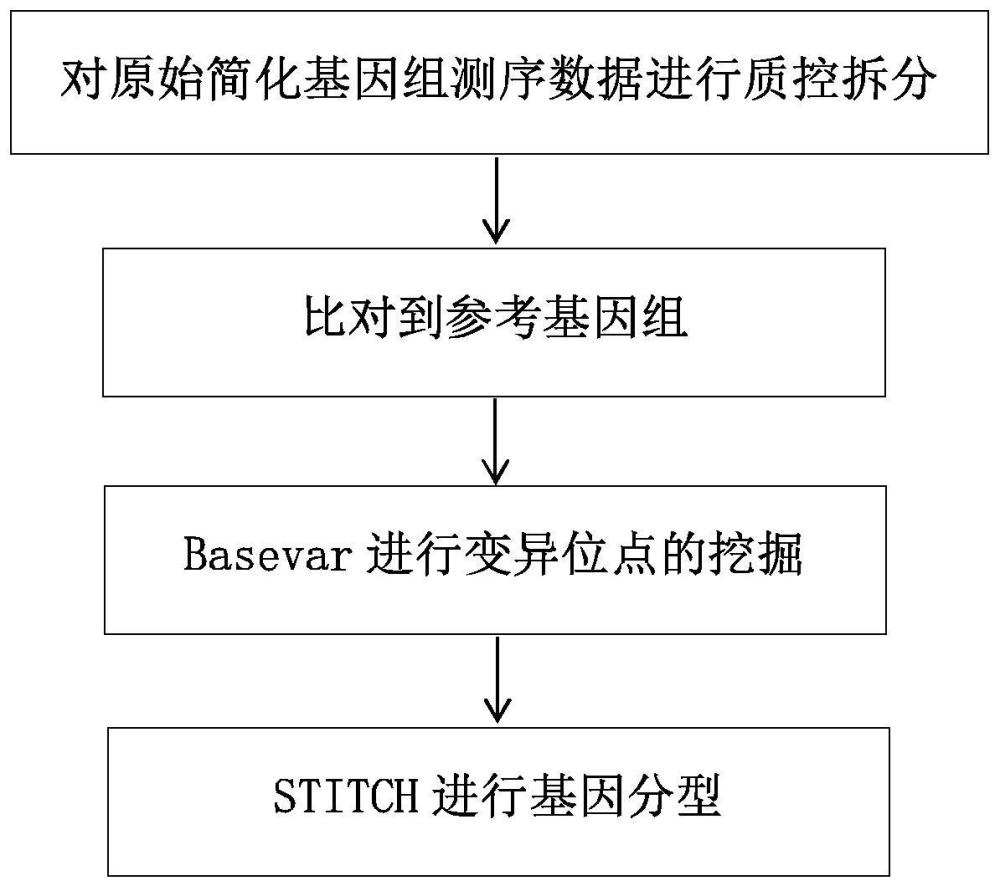
5、步骤2,将步骤1得到的定向基因组原始测序数据按照每个个体特定的barcode进行拆分,对拆分后个体的测序数据进行过滤,剔除不符合过滤标准的个体;
6、步骤3,将通过步骤2过滤筛选的个体的测序结果比对到参考基因组,对比对后的全部bam文件进行sort排序和构建索引;
7、步骤4,考察经步骤3对比后的全部bam文件所记载的基因组中每个位点是否为多态位点,得到各多态位点的位置信息;
8、步骤5,对步骤4得到的各多态位点进行进一步填充分型:通过em迭代算法处理所述育种群体的突变位点信息,输出该突变位点的基因分型结果。
9、进一步,步骤2的具体步骤为:
10、步骤2-1,对步骤1得到的定向基因组原始测序数据进行质控修剪,具体为:以4bp碱基为一个窗口,滑动窗口计算碱基平均质量,碱基质量阈值为15,如窗口内平均质量值低于阈值,则删除该窗口及之后的所有碱基;控制序列长度,剔除修剪后长度低于75bp的测序reads;
11、步骤2-2,将步骤1得到的定向基因组原始测序数据进行拆分,拆分时识别酶切位点序列的同时匹配每个个体特有的barcode;提取每条测序reads的barcode信息后,删除reads中的barcode序列;
12、步骤2-3,将经过步骤2-1质控修剪后的定向基因组原始测序数据,根据构建文库时的barcode序列记录进行个体拆分;
13、进一步,步骤4所述考察经步骤3对比后的产生的bam文件所记载的基因组中每个位点是否为多态位点,其考察过滤条件为:
14、剔除复等位基因、剔除eaf<0.01的位点、保留pcr≥0.5的位点。
15、上述eaf为估计的群体最小等位基因频率。
16、上述保留pcr≥0.5的位点指:对每一个候选多态位点的群体覆盖度(populationcoverage rate,pcr)进行评估过滤,具体为在该位点有reads覆盖(即该位点的测序乘数≥1)的样本数目占总样本数目的比例须大于等于0.5。
17、进一步,步骤5中所述每个多态位点的信息包括info score值,分型结果包括对上述info score值进行过滤处理的结果,对上述info score值进行过滤的标准为:次等位基因频率大于0.05、单个snp在群体中的call rate大于0.6、info_score大于0.4。
18、本发明所述的一种基于定向基因组测序获取全基因组snp位点的方法,其有益效果为:
19、本发明适用于定向基因组测序,建立了一种分析定向基因组测序数据多态位点的新流程,该方法避免了原有定向基因组测序流程中对测序深度敏感、分型准确度有限等不利因素,在低成本的基础上,可以更准确地得到分布于全基因组的、更高密度的snp信息。
20、此外,本过程得到的分型结果相较于原始定向基因组测序数据分型流程的结果,可以进一步向更高密度乃至全基因组水平填充,同时保持更高的填充准确性。
- 还没有人留言评论。精彩留言会获得点赞!