一种基于矩阵分解和异构图推理的病毒-药物关联预测方法
1.本发明涉及机器学习和生物信息技术领域,更具体地,涉及到一种基于受限玻尔兹曼机矩阵分解和异构图推理的病毒-药物关联预测方法。
背景技术:
2.人类与其他高等动物的生活与微生物群落密切相关,这些微生物群落包括细菌以及古细菌,病毒,真菌和原生动物。病毒是微生物中的一类,新病毒的爆发会对于人类造成巨大危害。比如,“新型冠状病毒”在全球迅速传播sars-cov-2引起了一种名为covid-19的严重急性呼吸综合征,目前尚未发现针对sars-cov-2的特殊疫苗或抗病毒药物。因此,尽快找到一种特殊的抗病毒药物来阻止sars-cov-2的传播是当务之急。此外,人类免疫缺陷病毒(hiv)感染经过这些阶段,包括病毒复制和传播、长时间的无症状阶段和cd4+t细胞的减少,可以引起获得性免疫缺陷综合症(aids)。埃博拉病毒(ebov)通过破损的皮肤或通过粘膜表面进入人体,导致ebov感染。ebov感染能够引起发烧、粘膜出血,甚至死亡。通过利用酸性内体和凝乳酶介导的内吞作用进行融合,寨卡病毒(zikv)可以感染细胞。zikv感染可以引起许多疾病,如登革热、黄热病和西尼罗河病毒。
3.通常,在感染病毒并患病后,人们首先使用药物来治疗疾病。因此,需要寻找有效的抗病毒药物。药物发现是制药科学的主要目标之一,是一个包括生物、化学、物理和统计等基础科学的跨学科研究领域。几千年来,大自然一直是医药产品的来源,许多有用的活性物质都是从植物来源中开发出来的。在20世纪,青霉素的发现是从微生物来源发现药物的起点。大多数药物是在细菌合成的天然产物的基础上,从先导结构开发出来的。源自细菌次级代谢产物的药物有多种用途,例如用于诊断、缓解或治疗或预防疾病或缓解不适。据估计,在微生物天然产物筛选的黄金时代(1940-1970),已经筛选了数千万种土壤微生物,这是一项巨大的努力,提供了当今已知的绝大多数微生物代谢物。这些物质包括广泛使用的抗菌疗法,如红霉素、链霉素、四环素、万古霉素和化疗药物,如阿霉素。当今诊所使用的所有抗生素中有90%来自微生物。目前,已知有23000多种具有抗菌活性的天然产物是由微生物产生的,而从植物和动物等高等生物中分离出来的天然产物只有25000种。
4.但是,目前药物开发面临两个主要挑战。一方面,一种药物时间周期长,从开始研发到市需要很长时间。另一方面,耐药性已经开始出现,对人类健康构成严重威胁。为了克服这一问题,组合化学已经被发展成为一项关键技术,能够产生大的筛选库,以满足高通量筛选的需要。此外,药物再利用,也称为药物重定位,是一种使用已获准上市的药物来治疗其他现有疾病的想法的方式。对于药物组合治疗和药物重新定位,确定药物与病毒的关联是至关重要的。因此,检测病毒与药物之间的相互作用对于病毒治疗学和药物开发具有重要意义。然而,传统的湿实验室实验(例如,基于培养的方法)发现病毒-药物的联系是费时、费力和昂贵的。因此,高效、准确地预测病毒-药物结合的计算方法是有限实验方法的有益补充。
5.总的来说,由于病毒耐药性问题、新药研制周期长,识别的病毒-药物关联对于药
物研制,疾病治疗具有重要意义。传统的实验方法耗时耗力,开发高效的计算方法来识别潜在的病毒-药物关联是一个极其迫切的问题。
技术实现要素:
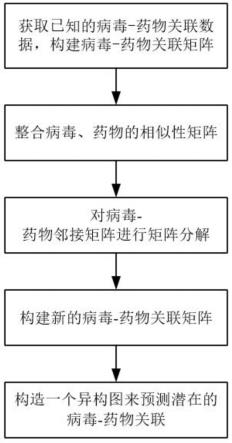
6.为了解决上面的问题,我们发明了一种基于矩阵分解和异构图推理的病毒-药物关联预测方法,更省时、省力的预测病毒-药物关联。
7.为了实现上述目的,本发明的技术方案:
8.一种基于矩阵分解和异构图推理的病毒-药物关联预测方法,包括步骤:
9.步骤一:获取已知的病毒-药物关联矩阵;
10.采用已知的病毒-药物数据组成数据集。数据的形式表示如下:
[0011][0012]
其中a(i,p)代表药物di与病毒v
p
是否相关,如果相关,则a(i,p)的值为1,否则为0。
[0013]
步骤二:整合病毒、药物的相似性矩阵;
[0014]
对于药物的相似性,将药物的化学结构相似性、药物的副作用相似性和药物的高斯交互作用谱核相似性整合起来,得到整合的药物相似性。如果药物有化学结构相似性或副作用相似性,则整合的药物相似性就是药物化学结构相似性和药物副作用相似性的平均值。否则,整合的药物相似性等于药物的高斯交互作用谱核相似性的值。其计算公式如下:
[0015][0016]
其中,ss1是药物化学结构相似性,ss2是药物副作用相似性,gd是药物的高斯相似性,sd是整合的药物相似性。
[0017]
对于病毒的相似性,将病毒序列相似性和病毒的高斯相互作用谱核相似性整合在一起,得到整合的病毒相似性。公式如下:
[0018][0019]
其中,mv是病毒的药物相似性,gv是病毒的高斯相似性,sv是整合的病毒相似性。
[0020]
步骤三:对病毒-药物邻接矩阵进行矩阵分解,并构建新的病毒-药物关联矩阵;
[0021]
由于数据集中药物-病毒邻接矩阵的某些关联可能是多余的或缺乏的,我们将已知的药物-病毒邻接矩阵分解为两部分。第一部分包括原始矩阵和低秩矩阵。低秩矩阵包括非冗余数据,可用于构建一个新的药物-病毒关联矩阵。第二部分是一个稀疏矩阵,其中的元素大部分为零。分解的公式如下。
[0022]
a=ax+e(3)
[0023]
其中,a为病毒药物关联矩阵,x为分解后的低秩矩阵,e分解后为稀疏矩阵;
[0024]
然后,使用核范数获得低秩矩阵x,用稀疏范数来获得稀疏矩阵e。上面方程(3)可以转化为如下方程:
[0025][0026]
其中:||
·
||
*
代表核范数,||
·
||
2,1
代表稀疏范数,α是用于控制权重;
[0027]
上面的公式(4)也可以等价表示为:
[0028][0029]
简单地说,上面的方程(5)可以被看作是一个约束和凸优化问题。打算采用不精确的增强拉格朗日乘法器(ialm)来解决这个问题。首先,将上述方程(5)转化为一个无约束问题。其次,采用如下公式(6)的增强拉格朗日函数使无约束问题最小化。
[0030][0031]
其中,y1、y2表示朗格朗日乘数;μ是一个惩罚系数;表示f范数;
[0032]
从上面的方程(6)中能得到两个解,它们分别被定义为x
*
和e
*
。然后,通过使用a和x
*
建立一个新的药物-病毒关联矩阵a
*
。其结果如下:
[0033]a*
=ax
*
ꢀꢀ
(7)
[0034]
步骤四:构造一个异构图来预测潜在的病毒-药物关联。
[0035]
将步骤三构建的新的药物-病毒关联矩阵和药物整合相似性、病毒整合相似性结合到一个异构图中。从异构图中预测药物与病毒潜在的关联概率。如果药物和病毒之间没有已知的关联,定义它们的潜在关联概率矩阵为公式如下:
[0036][0037]
其中,nv代表病毒的数量,nd代表药物的数量,v
l
代表第1至nv个病毒中的任意一个病毒,dm表示1至nd个药物中的任意一个药物。
[0038]
此外,异构图中边,即整合的药物相似性和综整合的病毒相似性的权重根据其端点的度数进行归一化处理。归一化的方程式如下所示:
[0039][0040][0041]
其中,v
l
代表第1至nv个病毒中的任意一个病毒,dm表示1至nd个药物中的任意一个药物;根据以前的研究,归一化过程有助于收敛。进一步,可以使用迭代方法来计算所有药物和所有病毒之间的潜在关联概率矩阵。迭代方程如下所示:
[0042]
p
k+1
=asv
×
pk×
sd+(1-a)a
*
ꢀꢀ
(11)
[0043]
其中,p
k+1
代表潜在关联概率矩阵的k+1次迭代,a代表惩罚系数。
[0044]
迭代结束后,所有药物和所有病毒之间的潜在关联概率矩阵即为预测的病毒-药物关联得分矩阵,得分矩阵仍然是一个nd行和nv列的矩阵,最后利用得分矩阵进行病毒-药物关联预测。
[0045]
与现有技术相比,本发明技术方案的有益效果是:
[0046]
本发明将病毒-药物关联预测问题作为预测任务,采用多源生物数据,包括已知的药物-病毒关联矩阵、药物化学结构相似性矩阵、药物副作用相似性矩阵、药物高斯相似性矩阵、病毒序列相似性矩阵、病毒高斯相似性矩阵,数据量丰富,有助于预测潜在的药物-病毒关联,实现了更加准确的病毒-药物的关联预测。
附图说明
[0047]
图1为本发明的方法流程图。
[0048]
图2为实施例所涉基于矩阵分解和异构图推理的病毒-药物关联预测方法和其它四种现有方法在数据集中的roc曲线和全局留一auc值比较图。
[0049]
图3为实施例所涉基于矩阵分解和异构图推理的病毒-药物关联预测方法和其它四种现有方法在数据集中的roc曲线和局部留一auc值比较图。
具体实施方式
[0050]
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,对于本领域技术人员来说,附图中某些公知结构及其说明可能省略是可以理解的。
[0051]
下面结合附图和实施例对本发明的技术方案做进一步的说明。
[0052]
本实例提供一种基于矩阵分解和异构图推理的病毒-药物关联预测方法(mdhgivda),如图1所示,包括以下步骤:
[0053]
步骤一:获取已知的病毒-药物关联矩阵。
[0054]
许多生物学实验发现了大量的病毒-药物关联。我们使用的病毒-药物关联信息来自drugvirus数据集。long等人基于drugvirus数据库构建了drugvirus数据集。drugvirus数据集包括933个已知的病毒-药物关联,其中包含175种药物和95种病毒。我们构建邻接矩阵a以存储病毒-药物关联信息。在a中,nd表示药物的数量,nm表示病毒的数量。如果药物与病毒有关,关联值为1,否则为0。
[0055][0056]
步骤二:整合病毒、药物的相似性矩阵;
[0057]
为了得到整合的药物相似性,我们将药物的化学结构相似性、药物的副作用相似性和药物的高斯交互作用谱核相似性整合起来。如果药物有化学结构相似性或副作用相似性,则整合的药物相似性就是药物化学结构相似性和药物副作用相似性的平均值。否则,整合的药物相似性等于药物的高斯交互作用谱核相似性的值。其计算公式如下:
[0058][0059]
其中,ss1是药物化学结构相似性,ss2是药物副作用相似性,gd是药物的高斯相似性,sd是整合的药物相似性。
[0060]
对于病毒相似性,我们将病毒序列相似性和病毒的高斯相互作用谱核相似性整合在一起,得到整合的病毒相似性。公式如下:
[0061][0062]
其中是病毒的药物相似性,是病毒的高斯相似性,是整合的病毒相似性。
[0063]
步骤三:对病毒-药物邻接矩阵进行矩阵分解,并构建新的病毒-药物关联矩阵;
[0064]
由于数据集中药物-病毒邻接矩阵的某些关联可能是多余的或缺乏的,我们将已知的药物-病毒邻接矩阵分解为两部分。第一部分包括原始矩阵和低秩矩阵。低秩矩阵包括非冗余数据,可用于构建一个新的药物-病毒关联矩阵。第二部分是一个稀疏矩阵,其中的元素大部分为零。分解的公式如下。
[0065]
a=ax+e
[0066]
然后,使用核范数获得低秩矩阵x,用稀疏范数来获得稀疏矩阵e。这样一来,上面方程可以转化为如下方程:
[0067][0068]
其中:||
·
||
*
代表核范数,||
·
||
2,1
代表稀疏范数,α是用于控制权重;
[0069]
上面的公式也可以等价表示为:
[0070][0071]
简单地说,上面的方程可以被看作是一个约束和凸优化问题。打算采用不精确的增强拉格朗日乘法器(ialm)来解决这个问题。首先,可以将上述方程转化为一个无约束问题。其次,采用如下的增强拉格朗日函数使无约束问题最小化。
[0072][0073]
我们可以从上面的方程中得到两个解,它们分别被定义为x
*
和e
*
。然后,通过使用a和x
*
建立一个新的药物-病毒关联矩阵a
*
。其结果如下:
[0074]a*
=ax
*
[0075]
步骤四:构造一个异构图来预测潜在的病毒-药物关联。
[0076]
将新的药物-病毒关联矩阵和药物整合相似性、病毒整合相似性结合到一个异构图中。然后,可以从异构图中预测药物与病毒潜在的关联概率。如果药物和病毒之间没有已知的关联,我们定义它们的潜在关联概率为公式如下:
[0077][0078]
此外,边(整合的药物相似性和综整合的病毒相似性)的权重根据其端点的度数进行归一化处理。归一化的方程式如下所示:
[0079][0080]
[0081]
根据以前的研究,归一化过程有助于收敛。进一步,可以使用迭代方法来计算药物和病毒之间的潜在关联概率。迭代方程如下所示:z是分区函数,表示如下:
[0082]
p
k+1
=asv
×
pk×
sd+(1-a)a
*
[0083]
最后,预测的病毒-药物关联得分矩阵是一个nd行和nv列的矩阵。我们使用p来保这个得分矩阵。根据预测的分数进行排序,分数越高表明病毒-药物关联的可能性越大。并且,根据排序的结果,可以给出某种病毒和药物之间的关联可能性排名,预测关联性具有很大的参考价值,有助于生物医学领域针对性地研究某种病毒和某种药物之间相互作用关系,从而为有助于研发药物,治疗疾病。
[0084]
评估方法:我们采用全局loocv、局部loocv和五折交叉验证法对本发明提出的方法的预测性能进行评估。在loocv中,依次选择每个已知的病毒-药物关联作为测试样本,其余已知的病毒-药物关联则作为训练样本。对于全局loocv,所有未知的病毒-药物对都被用作候选样本。然后,我们用训练样本来训练模型,用训练好的模型来预测测试样本和候选样本的得分。我们进一步根据全局loocv的预测分数对测试样本和候选样本进行排名。最后,我们得到了所有测试样本的排名。而在局部loocv中,测试样品的得分与包括测试样品中被调查药物在内的候选样品的得分进行排名。最后,我们还得到了所有测试样本的排名。在五折交叉验证中,已知的病毒-药物关联被随机分为五个子集,每个子集依次被视为测试样本,其他四个子集被视为训练样本。所有未知的病毒-药物对将被视为候选样本。随后,我们将每个测试样本的得分与候选样本的得分进行排名。最后,我们得到了所有测试样本的排名。为了避免随机样本划分造成的偏差,五折交叉验证被重复了100次。此外,我们绘制了roc曲线。并计算auc曲线下的面积auc来评估方法的预测性能。
[0085]
评估结果:对于五折交叉验证,我们的方法(mdhgivda)获得的auc和标准差是0.8299+/-0.0037,对比方法hgimda、imcmda、katcmda、rlsmda对应的结果分别是0.6996+/-0.0022、0.6808+/-0.0040、0.8228+/-0.0023、0.6513+/-0.0229。在全局留一中交叉验证中,结果如图二所示,mdhgivda取得的auc是0.8528,高于hgimda的0.7084、imcmda的0.6902、katcmda的0.8247、rlsmda的0.6849。在局部留一交叉验证中,结果如图三所示,mdhgivda取得的auc是0.8532,高于hgimda的0.7537、imcmda的0.7436、katcmda的0.8247、rlsmda的0.6815。
[0086]
案例研究:进一步地,我们使用案例研究来进一步评估我们的方法(mdhgivda)的预测性能。我们选择了三种病毒作为代表来实现案例研究,三种病毒分别是寨卡病毒、新冠病毒、艾滋病病毒1型。通过实施mdhgivda,我们预测三种病毒相关联的药物。然后,我们根据预测得分对相关药物进行排名,并通过在pubmed上搜索文献验证了三种病毒前10个潜在相关药物。结果结果如表一、表二、表三所示。对于寨卡病毒、新冠病毒、艾滋病病毒1型,预测的前十个药物中,分别有10、8、8个被验证。
[0087]
表一:预测的寨卡病毒前十个相关的药物
[0088]
[0089][0090]
表二:预测的新冠病毒前十个相关的药物
[0091]
[0092][0093]
表三:预测的艾滋病病毒1型前十个相关的药物
[0094]
[0095][0096]
相同或相似的标号对应相同或相似的部件;
[0097]
附图中描述位置关系的用语仅用于示例性说明,不能理解为对本专利的限制;
[0098]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求
的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1