一种PEG化喜树碱长效缓释凝胶的制作方法
一种peg化喜树碱长效缓释凝胶
技术领域
1.本发明属于医药化工领域,具体涉及一种peg化喜树碱衍生物及其与多氨基化合物交联形成的长效缓释凝胶。
背景技术:
2.喜树碱由1966年美国的monroe e.wall博士从喜树的皮中分离得到,其在抗肿瘤活性上的优异表现引起了人们的广泛关注。它可以选择性抑制拓扑异构酶ⅰ,与topo
ⅰ‑
dna形成的复合物结合,稳定此复合物,从而使断裂的dna链不能重新接合,阻止dna复制及rna合成,为细胞周期s期特异性药物。另外,它还能直接破坏dna结构。实验证明,喜树碱对多种动物肿瘤有抑制作用,与常用抗肿瘤药物无交叉耐药。但是,其分子结构中喹琳环上氮的特殊碱性导致其水溶性差,不能直接用于人体的非肠道给药。同时由于喜树碱副作用大,体内半衰期短,体内残留时间长等缺点,使得其应用受到一定的限制。
3.与以往的常规剂型如片剂、胶囊、注射剂相比,缓释、控释制剂能够减少给药次数,改善患者的顺应性。尤其针对的是半衰期短的或者需要频繁给药的药物,可以减少服药次数,使药物浓度平稳,避免峰谷现象,有利于降低药物的毒副作用,增加药物治疗的稳定性和安全性。同时可以减少用药的总量,用较少的剂量达到更好的效果。
4.现有技术通过物理包埋或化学修饰方式,增强喜树碱的水溶性,延长消除时间,起到降低毒性提高药效的作用。
5.cn101352420b通过膜乳化法制备了装载羟基喜树碱的缓释微球,该专利制备的缓释微球能通过微导管直接在瘤体内注射方式给药,可实现靶向给药,降低枪击喜树碱的全身毒副作用,可以在体内存留30天左右。但其制备过程需要用到有机溶剂,对人体存在一定的刺激性。
6.cn106236699b介绍了“溶液喷涂”或“热熔挤出”工艺两种制备装载10-羟基喜树碱的体内植入物的制备方法,通过该方法制备植入剂能在体内缓慢释放hcpt,增强hcpt的药物作用,降低不良反应。同时植入剂能够直接与肿瘤接触,增加了肿瘤局部的药物浓度,降低了血中的药物浓度。但是该发明描述制备工艺复杂,且无法如微球药剂一样通过注射的方法植入体内,药物释放量较少,实际使用效果不理想。
7.cn101156854a通过将喜树碱及其衍生物组装在不同高分子层数的微胶囊中,以达成药物缓释的效果。该发明工艺简单,使用材料均为可降解材料,制备过程不存在化学反应,对人体影响较小。但该药剂在15小时左右就可以达到40%的药物释放率,药物释放太快,容易引起药物毒副作用。
8.相比于喜树碱药物,peg化喜树碱注射液,可以显著提高药物的水溶性,实现体内较长时间的循坏。临床试验显示,peg化喜树碱在体内的半衰期可以达到77小时左右(rowinsky ek等人,j clin oncol 21:148
–
157)。然而临床实验中,仍然需要多次给药以维持足够的药物浓度,而且全身给药会给健康器官带来较大的全身毒性。通过物理包埋凝胶可实现局部给药,以提高药物在肿瘤局部的浓度来降低药物的全身毒性,然而对于小分子
喜树碱药物来说,药物从凝胶中的释放速率较快,难以达到长期缓释的目的,例如喜树碱在琼脂凝胶的释放周期仅为两个小时左右(j.liu et al./european polymer journal 42(2006))。因此,为了提高喜树碱药物的抗肿瘤能力,迫切需要一种能在肿瘤局部长期缓释药物的方法。
技术实现要素:
9.本发明针对现有技术不足,提供了一种peg化喜树碱衍生物以及该peg化喜树碱衍生物与多氨基化合物原位交联形成的可注射性凝胶。可随着凝胶的降解实现药物缓慢释放,达到长期缓释药物的目的。
10.本发明具体技术方案如下:
11.一种peg化喜树碱衍生物,具有如下结构:其中n代表1~10的正整数,优选2~6的正整数。peg为醛基封端的星形多臂聚乙二醇。
12.优选的,所述醛基封端的星形多臂聚乙二醇的臂数为2-8,单臂分子量1000-5000da。
13.所述醛基选自芳香醛、烷基醛中的一种或几种,所述醛基与星形多臂聚乙二醇之间以酯键、醚键、酰胺键、氨酯键、亚胺键或脲键化学键连接,优选酰胺键或酯键连接。
14.本发明另一目的在于提供一种peg化喜树碱缓释凝胶,由本发明所述的peg化喜树碱衍生物和多氨基化合物原位交联而成。
15.优选的,所述多氨基化合物中氨基与peg化喜树碱衍生物中醛基的摩尔比为0.4~4.4:1,更优选1~4:1;所述多氨基化合物为聚赖氨酸或聚赖氨酸与聚乙烯亚胺的混合物,聚赖氨酸与聚乙烯亚胺的摩尔比为2~30:3,更优选2~10:3。
16.本发明所述的peg化喜树碱缓释凝胶,采用如下方法制备而成:将peg化喜树碱衍生物溶解在ph4-6缓冲液中,配制溶液;将多氨基化合物溶解在ph4-10缓冲液中,配制多氨基化合物溶液;将两者混合得到peg化喜树碱缓释凝胶。
17.所述peg化喜树碱衍生物溶液的质量体积百分浓度为2-30%,优选,10~20%;所述多氨基化合物溶液质量体积百分浓度为0.5-20%,优选1-5%。
18.本发明另一目的在于提供本发明所述的peg化喜树碱衍生物或peg化喜树碱在制备抗癌缓释药物中的应用。
19.本发明优点:
20.本发明通过可降解连接键将喜树碱偶联在peg上,同时通过peg端基功能化进一步与多氨基化合物化学偶联,实现原位凝胶。本发明所述peg化喜树碱凝胶具有长效缓释作用,显著减少药物突释,药物随降解而缓慢释放,周期长达一到三个月。
附图说明
21.图1为不同桥接链长的peg化喜树碱凝胶的药物缓释曲线。
22.图2为不同取代率的peg化喜树碱凝胶的药物缓释曲线。
23.图3为不同取代率的peg化喜树碱凝胶的在37℃的溶胀曲线。
具体实施方式
24.以下通过实施例说明本发明的具体步骤,但不受实施例限制。在本发明中所使用的术语,除非另有说明,一般具有本领域普通技术人员通常理解的含义。
25.下面结合具体实施例并参照数据进一步详细描述本发明。应理解,这些实施例只是为了举例说明本发明,而非以任何方式限制本发明的范围。在以下实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。
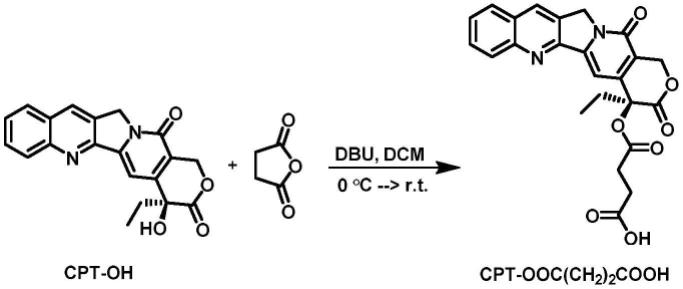
26.实施例1 cpt-ooc(ch2)2cooh的合成
[0027][0028]
称取喜树碱(cpt-oh,174mg)、丁二酸酐(150mg)于50ml茄瓶中,再加入1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu,0.23ml),二氯甲烷(dcm)15ml,室温反应4h。加入盐酸水溶液猝灭反应,再加入大量二氯甲烷萃取,柱层析分离,得浅黄色粉末状固体114mg。1h nmr(400mhz,dmso-d6,)δ12.17(s,1h),8.69(s,1h),8.18(d,j=8.5hz,1h),8.13(d,j=8.2hz,1h),7.91
–
7.82(m,1h),7.72(t,j=7.5hz,1h),7.13(s,1h),5.48(s,2h),5.30(s,2h),2.84
–
2.67(m,2h),2.47(m,2h),2.15(q,j=7.4hz,2h),0.91(t,j=7.4hz,3h).hrms(positive esi)m/z found(calcd for c
24h20
n2o7+h
+
):449.1344(449.1343).hrms(positive esi)m/z found(calcd for c
24h20
n2o
7-h
+
):447.1198(447.1198)。
[0029]
实施例2 cpt-ooc(ch2)3cooh的合成
[0030][0031]
称取喜树碱(cpt-oh,174mg)、戊二酸酐(171mg)于50ml茄瓶中,再加入1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu,0.23ml),二氯甲烷(dcm)15ml,室温反应4h。加入盐酸水溶液猝灭反应,再加入大量二氯甲烷萃取,柱层析分离,得浅黄色粉末状固体85mg。1h nmr(400mhz,dmso-d6,)δ12.00(s,1h),8.66(s,1h),8.16
–
8.08(m,2h),7.83(ddd,j=8.4,6.8,
found(calcd for c
27h26
n2o7+h
+
):491.1812(491.1813)。
[0038]
实施例5 cpt-ooc(ch2)6cooh的合成:
[0039][0040]
称取喜树碱(cpt-oh,174mg)、辛二酸(261mg)、4-二甲氨基吡啶(dmap,183mg)、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edci,576mg)于50ml茄瓶中,再加入二氯甲烷(dcm)15ml,室温反应12h。加入盐酸水溶液猝灭反应,再加入大量二氯甲烷萃取,柱层析分离,得浅黄色粉末状固体96mg。1h nmr(400mhz,dmso-d6)δ11.88(s,1h),8.66(s,1h),8.11(t,j=9.1hz,2h),7.83(t,j=7.7hz,1h),7.68(t,j=7.5hz,1h),7.01(s,1h),5.45(s,2h),5.27(s,2h),2.50(m,2h),2.11(m,4h),1.52(q,j=7.3hz,2h),1.41(t,j=7.3hz,2h),1.26(m,4h),0.88(t,j=7.4hz,3h).hrms(positive esi)m/z found(calcd for c
28h28
n2o7+h
+
):505.1969(505.1969)。
[0041]
表1为实施例1-5具有不同连接键长的喜树碱衍生物的最大紫外吸收波长及吸光系数,结果表明对喜树碱的修饰未影响其它官能团结构。
[0042]
表1 cpt-ooc(ch2)ncooh的紫外吸收统计
[0043][0044]
实施例6 peg-(oh)7(cpt)1的合成
[0045]
称量1.00g peg(oh)8(八臂星形聚乙二醇,单臂分子量1875da)原料,分别将实施例1-5合成的喜树碱二酸酐衍生物,0.311g 1-乙基-(3-二甲基氨基丙基)碳酰二亚胺(edc)和0.294g 4-二甲氨基吡啶加入到30ml二氯甲烷(dcm)中,常温搅拌12h。将反应溶液稀释于250ml dcm中,用250ml氯化钠水溶液水洗三次,接着浓缩后在乙醚中沉淀。最后,将得到的固体样品真空抽干称重得到产物。
[0046]
实施例7 peg-(p-phcho)7(ooc(ch2)2coocpt)1的合成
[0047]
将1.20g实施例6制得的peg-(oh)7(ooc(ch2)2coocpt)1,0.217g对甲酰基苯甲酸,0.374g 1-乙基-(3-二甲基氨基丙基)碳酰二亚胺(edc)和0.353g 4-二甲氨基吡啶加入到30ml二氯甲烷(dcm)中,常温搅拌12h。将反应溶液稀释于250ml dcm中,用250ml氯化钠水溶液水洗三次,接着浓缩后在乙醚中沉淀。最后,将得到的固体样品真空抽干称重得0.835g产物。
[0048]
实施例8 peg-(p-phcho)7(ooc(ch2)3coocpt)1的合成
[0049]
将1.20g实施例6制得的peg-(oh)7(ooc(ch2)3coocpt)1,0.217g对甲酰基苯甲酸,,0.374g 1-乙基-(3-二甲基氨基丙基)碳酰二亚胺(edc)和0.353g 4-二甲氨基吡啶加入到30ml二氯甲烷(dcm)中,常温搅拌12h。将反应溶液稀释于250ml dcm中,用250ml氯化钠水溶液水洗三次,接着浓缩后在乙醚中沉淀。最后,将得到的固体样品真空抽干称重得0.735g产物。
[0050]
实施例9 peg-(p-phcho)7(ooc(ch2)4coocpt)1的合成
[0051]
将1.653g实施例6制得的peg-(oh)7(ooc(ch2)4coocpt)1,0.298g对甲酰基苯甲酸,0.515g 1-乙基-(3-二甲基氨基丙基)碳酰二亚胺(edc)和0.486g 4-二甲氨基吡啶加入到30ml二氯甲烷(dcm)中,常温搅拌12h。将反应溶液稀释于250ml dcm中,用250ml氯化钠水溶液水洗三次,接着浓缩后在乙醚中沉淀。最后,将得到的固体样品真空抽干称重得1.234g产物。
[0052]
实施例10 peg-(p-phcho)7(ooc(ch2)5coocpt)1的合成
[0053]
将1.587g实施例6制得的peg-(oh)7(ooc(ch2)5coocpt)1,0.287g对甲酰基苯甲酸,,0.494g 1-乙基-(3-二甲基氨基丙基)碳酰二亚胺(edc)和0.466g 4-二甲氨基吡啶加入到30ml二氯甲烷(dcm)中,常温搅拌12h。将反应溶液稀释于250ml dcm中,用250ml氯化钠水溶液水洗三次,接着浓缩后在乙醚中沉淀三次。最后,将得到的固体样品真空抽干称重得0.922g产物。
[0054]
实施例11 peg-(p-phcho)7(ooc(ch2)6coocpt)1的合成
[0055]
将1.498g实施例6制得的peg-(oh)7(ooc(ch2)6coocpt)1,0.275g对甲酰基苯甲酸,0.466g 1-乙基-(3-二甲基氨基丙基)碳酰二亚胺(edc)和0.440g 4-二甲氨基吡啶加入到30ml二氯甲烷(dcm)中,常温搅拌12h。将反应溶液稀释于250ml dcm中,用250ml氯化钠水溶液水洗三次,接着浓缩后在乙醚中沉淀三次。最后,将得到的固体样品真空抽干称重得0.967g产物。
[0056]
实施例12 peg-(oh)6(ooc(ch2)3coocpt)2的合成
[0057]
反应方程式:
[0058]
[0059]
将cpt-ooc(ch2)3cooh(206.8mg)、peg(oh)8(3.89g)、edci(4.887g)和dmap(2.926g),放入圆底烧瓶中,加入二氯甲烷溶剂30ml,反应室温搅拌过夜。然后萃取水洗3次,并在乙醚中沉淀。最终得到产物2.9g。
[0060]
其中,化学位移为δ8.7,8.15,7.87,7.72,7.06为cpt芳基氢,δ5.50,5.31为cpt中亚甲基ch2的氢,δ2.15,0.92为cpt中ch2ch3的氢,δ2.60,2.40,1.79为cpt-ooc(ch2)3coo的ch2ch2ch2的氢,δ4.59为peg羟基的峰,δ4.11为peg中与羧基酯化生成的cooch2的氢。
[0061]
实施例13 peg-(p-phcho)6(ooc(ch2)3coocpt)2的合成
[0062]
将2.059g实施例12制得的喜树碱衍生物peg-(oh)6(ooc(ch2)3coocpt)2,0.588g对甲酰基苯甲酸(p-cba),1.009g 1-乙基-(3-二甲基氨基丙基)碳酰二亚胺(edc)和0.962g4-二甲氨基吡啶加入到二氯甲烷(dcm)中,搅拌12h。将反应溶液稀释于约250ml dcm中,用水洗三次,旋蒸浓缩后在乙醚中沉淀,最后,将得到的固体样品真空抽干称重得0.880g产物。
[0063]
实施例14 peg-(oh)4(ooc(ch2)3coocpt)4的合成
[0064]
反应方程式:
[0065][0066]
将cpt-ooc(ch2)3cooh(415.8mg,)、peg(oh)8(3.99g,)、edci(7.668g,)和dmap(5.8664g,48mmol),放入圆底烧瓶中,加入二氯甲烷溶剂30ml,反应室温搅拌过夜。然后萃取水洗3次,并在乙醚中沉淀。最终得到产物3.6g。
[0067]
其中,化学位移为δ8.7,8.15,7.87,7.72,7.06为cpt芳基氢,δ5.50,5.31为cpt中亚甲基ch2的氢,δ2.15,0.92为cpt中ch2ch3的氢,δ2.60,2.40,1.79为cpt-ooc(ch2)3coo的ch2ch2ch2的氢,δ4.59为peg羟基的峰,δ4.11为peg中与羧基酯化生成的cooch2的氢。
[0068]
实施例15 peg-(p-phcho)4(ooc(ch2)3coocpt)4的合成
[0069]
将2.035g实施例14制得的喜树碱衍生物peg-(oh)4(ooc(ch2)3coocpt)4、0.370g对甲酰基苯甲酸(p-cba),0.666g 1-乙基-(3-二甲基氨基丙基)碳酰二亚胺(edc)和0.634g4-二甲氨基吡啶加入到二氯甲烷(dcm)中,搅拌12h。将反应溶液稀释于约250ml dcm中,用水洗三次,旋蒸浓缩后在乙醚中沉淀,最后,将得到的固体样品真空抽干5h后称重得0.880g产物。
[0070]
表2为实施例7~11和实施例15偶联苯醛基及喜树碱的peg的h nmr关键化学位移,从化学位移比例关系确认了取代率,该工艺能精准的调控药物及交联官能团的比例。
[0071]
表2 peg-(p-phcho)n(ooc(ch2)ncoocpt)n的h nmr结果统计
[0072]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1