一种蛋白质相互作用的预测方法、系统、设备以及介质与流程
1.本发明涉及蛋白质领域,具体涉及一种蛋白质相互作用的预测方法、系统、设备以及存储介质。
背景技术:
2.蛋白质是由一个或多个α-氨基酸残基长链条组成的生物大分子,具有四级结构水平:一级结构、二级结构、三级结构和四级结构,其中三级结构和四级结构具有复杂的空间结构,具有功能性。蛋白质在执行其生化功能时,总会通过非共价键形成蛋白质复合体(protein complex),这些不同蛋白质之间的相互作用(protein-protein interaction,ppi)构成了细胞生化反应网络的一个主要组成部分,在绝大多数生化功能中扮演极重要的角色。
3.根据模型输入的特征,ppi预测方法主要分为:基于序列和基于蛋白质三维结构这两类。其中基于序列的方法主要有:三元组(conjoint triads,ct)、自协方差(auto-covariance,ac)方法。
4.但是基于传统实验识别蛋白质相互作用的技术存在费时、覆盖度有限且价格昂贵的缺点。近年来,研究者已经研发了一些利用cnn或者lstm和蛋白质氨基酸序列识别蛋白质相互作用的方法。但这些方法普遍存在一些缺点:蛋白质氨基酸序列向量化编码方法无法充分提取互作特征,并且忽视多种氨基酸序列编码和分类器间的互补信息,同时还存在未能充分利用三维结构信息进行预测的问题。
技术实现要素:
5.有鉴于此,为了克服上述问题的至少一个方面,本发明实施例提出一种蛋白质相互作用的预测方法,包括以下步骤:
6.s1,获取待预测的蛋白质的三维结构和氨基酸序列;
7.s2,将所述蛋白质的三维结构放置在体素为l*l*l的三维空间中,其中,所述蛋白质的三维结构的质心与所述三维空间的原点重合,并将所述蛋白质的三维结构的所有坐标进行缩放;
8.s3,根据所述蛋白质的三维结构是否穿过所述三维空间中的体素得到l*l*l的形状向量,其中,若所述蛋白质的三维结构穿过体素,则值为1,否则为0;并根据所述蛋白质的三维结构中每一个氨基酸在所述三维空间的位置以及疏水性和侧链净电荷指数得到l*l*l的第一特性向量和l*l*l的第二特性向量;
9.s4,利用所述氨基酸序列生成第三特征向量;
10.s5,将所述形状向量、所述第一特征向量、所述第二特征向量以及所述第三特征向量进行连接得到最终的特征向量;
11.s6,将所述最终的特征向量输入到训练后的预测模型中进行预测以得到待预测的蛋白质的相互作用。
12.在一些实施例中,利用所述氨基酸序列生成第三特征向量,进一步包括基于下式得到向量中的每一个元素值:
[0013][0014]
其中,p表示蛋白质的氨基酸序列,长度为nr,m是序列中第m个氨基酸的编号,n是理化特性的编号,lag是滞后的数值,p
m,n
为第m个氨基酸的编号为n的理化特性,从而将氨基酸序列转换为(n
×
lag)维度的向量。
[0015]
在一些实施例中,将所述蛋白质的三维结构的所有坐标进行缩放,进一步包括:
[0016]
利用对所述蛋白质的三维结构的所有坐标进行缩放,其中,所述r
max
为所述三维空间中最大内切球半径。
[0017]
在一些实施例中,还包括:
[0018]
利用连接层、线性层、h个transformer模块以及归一化层构建预测模型;
[0019]
构建训练集,并对所述训练集中的每一个样本进行步骤s2-s5的处理以得到每一个样本对应的最终的特征向量;
[0020]
利用每一个样本对应的最终的特征向量对所述预测模型进行训练。
[0021]
基于同一发明构思,根据本发明的另一个方面,本发明的实施例还提供了一种蛋白质相互作用的预测系统,包括:
[0022]
获取模块,配置为获取待预测的蛋白质的三维结构和氨基酸序列;
[0023]
缩放模块,配置为将所述蛋白质的三维结构放置在体素为l*l*l的三维空间中,其中,所述蛋白质的三维结构的质心与所述三维空间的原点重合,并将所述蛋白质的三维结构的所有坐标进行缩放;
[0024]
第一生成模块,配置为根据所述蛋白质的三维结构是否穿过所述三维空间中的体素得到l*l*l的形状向量,其中,若所述蛋白质的三维结构穿过体素,则值为1,否则为0;并根据所述蛋白质的三维结构中每一个氨基酸在所述三维空间的位置以及疏水性和侧链净电荷指数得到l*l*l的第一特性向量和l*l*l的第二特性向量;
[0025]
第二生成模块,配置为利用所述氨基酸序列生成第三特征向量;
[0026]
连接模块,配置为将所述形状向量、所述第一特征向量、所述第二特征向量以及所述第三特征向量进行连接得到最终的特征向量;
[0027]
预测模块,配置为将所述最终的特征向量输入到训练后的预测模型中进行预测以得到待预测的蛋白质的相互作用。
[0028]
在一些实施例中,第二生成模块还配置为:
[0029][0030]
其中,p表示蛋白质的氨基酸序列,长度为nr,m是序列中第m个氨基酸的编号,n是理化特性的编号,lag是滞后的数值,p
m,n
为第m个氨基酸的编号为n的理化特性,从而将氨基
酸序列转换为(n
×
lag)维度的向量。
[0031]
在一些实施例中,缩放模块还配置为:
[0032]
利用对所述蛋白质的三维结构的所有坐标进行缩放,其中,所述r
max
为所述三维空间中最大内切球半径。
[0033]
在一些实施例中,还包括神经网络模块,配置为:
[0034]
利用连接层、线性层、h个transformer模块以及归一化层构建预测模型;
[0035]
构建训练集,并对所述训练集中的每一个样本进行步骤s2-s5的处理以得到每一个样本对应的最终的特征向量;
[0036]
利用每一个样本对应的最终的特征向量对所述预测模型进行训练。
[0037]
基于同一发明构思,根据本发明的另一个方面,本发明的实施例还提供了一种计算机设备,包括:
[0038]
至少一个处理器;以及
[0039]
存储器,所述存储器存储有可在所述处理器上运行的计算机程序,其特征在于,所述处理器执行所述程序时执行如上所述的任一种蛋白质相互作用的预测方法的步骤。
[0040]
基于同一发明构思,根据本发明的另一个方面,本发明的实施例还提供了一种计算机可读存储介质,所述计算机可读存储介质存储有计算机程序,所述计算机程序被处理器执行时执行如上所述的任一种蛋白质相互作用的预测方法的步骤。
[0041]
本发明具有以下有益技术效果之一:本发明提出的方案能够充分利用蛋白质不同维度的信息:一维序列信息和三维结构信息进行蛋白质相互作用预测的模型。
附图说明
[0042]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的实施例。
[0043]
图1为本发明的实施例提供的蛋白质相互作用的预测方法的流程示意图;
[0044]
图2本发明的实施例提供的r
max
和v之间关系的示意图;
[0045]
图3本发明的实施例提供的20种氨基酸分为五大类示意图;
[0046]
图4本发明的实施例提供的20种氨基酸的6大理化特性示意图;
[0047]
图5本发明的实施例提供的特征提取流程示意图;
[0048]
图6本发明的实施例提供的预测模型示意图;
[0049]
图7为本发明的实施例提供的蛋白质相互作用的预测系统的结构示意图;
[0050]
图8为本发明的实施例提供的计算机设备的结构示意图;
[0051]
图9为本发明的实施例提供的计算机可读存储介质的结构示意图。
具体实施方式
[0052]
为使本发明的目的、技术方案和优点更加清楚明白,以下结合具体实施例,并参照附图,对本发明实施例进一步详细说明。
[0053]
需要说明的是,本发明实施例中所有使用“第一”和“第二”的表述均是为了区分两个相同名称非相同的实体或者非相同的参量,可见“第一”“第二”仅为了表述的方便,不应理解为对本发明实施例的限定,后续实施例对此不再一一说明。
[0054]
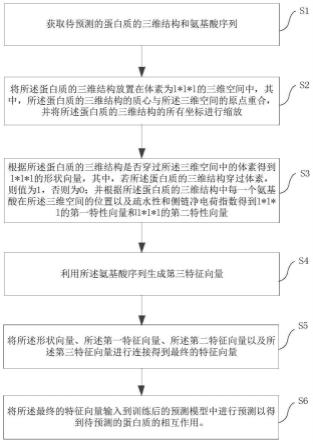
根据本发明的一个方面,本发明的实施例提出一种蛋白质相互作用的预测方法,如图1所示,其可以包括步骤:
[0055]
s1,获取待预测的蛋白质的三维结构和氨基酸序列;
[0056]
s2,将所述蛋白质的三维结构放置在体素为l*l*l的三维空间中,其中,所述蛋白质的三维结构的质心与所述三维空间的原点重合,并将所述蛋白质的三维结构的所有坐标进行缩放;
[0057]
s3,根据所述蛋白质的三维结构是否穿过所述三维空间中的体素得到l*l*l的形状向量,其中,若所述蛋白质的三维结构穿过体素,则值为1,否则为0;并根据所述蛋白质的三维结构中每一个氨基酸在所述三维空间的位置以及疏水性和侧链净电荷指数得到l*l*l的第一特性向量和l*l*l的第二特性向量;
[0058]
s4,利用所述氨基酸序列生成第三特征向量;
[0059]
s5,将所述形状向量、所述第一特征向量、所述第二特征向量以及所述第三特征向量进行连接得到最终的特征向量;
[0060]
s6,将所述最终的特征向量输入到训练后的预测模型中进行预测以得到待预测的蛋白质的相互作用。
[0061]
本发明提出的方案能够充分利用蛋白质不同维度的信息:一维序列信息和三维结构信息进行蛋白质相互作用预测的模型。
[0062]
在一些实施例中,将所述蛋白质的三维结构的所有坐标进行缩放,进一步包括:
[0063]
利用对所述蛋白质的三维结构的所有坐标进行缩放,其中,所述r
max
为所述三维空间中最大内切球半径。
[0064]
具体的,对于三维结构信息,对蛋白质结构的三维坐标进行如下操作:将蛋白质结构的质心移动到原点(立方体v的中心),对所有坐标值进行缩放,乘数
[0065]
其中,本工作中r
max
=40,l=32,r
max
和v的关系如图2所示。
[0066]
在一些实施例中,步骤s3,根据所述蛋白质的三维结构是否穿过所述三维空间中的体素得到l*l*l的形状向量,其中,若所述蛋白质的三维结构穿过体素,则值为1,否则为0;并根据所述蛋白质的三维结构中每一个氨基酸在所述三维空间的位置以及疏水性和侧链净电荷指数得到l*l*l的第一特性向量和l*l*l的第二特性向量,具体的,使用volumetric representation,将蛋白质的三维坐标表示成在固定三维空间中体素表示(网格密度为l*l*l),如果蛋白质的骨架穿过体素,则值为1,否则值为0。但体素表示只包含了蛋白质三维结构中的形状,除了蛋白质三维结构的形状,蛋白质中氨基酸的疏水性和侧链净电荷指数也从根本上影响了蛋白质之间的相互作用,因此本工作也将提取这部分性质,同时作为模型的输入。同样使用上述volumetric representation,将蛋白质中氨基酸的疏水性和侧链净电荷指数分别用三维立方空间v中体素表示。
[0067]
这样,三维结构信息通过volumetric representation生成三个l*l*l的向量。
[0068]
在一些实施例中,利用所述氨基酸序列生成第三特征向量,进一步包括基于下式得到向量中的每一个元素值:
[0069][0070]
其中,p表示蛋白质的氨基酸序列,长度为nr,m是序列中第m个氨基酸的编号,n是理化特性的编号,lag是滞后的数值,p
m,n
为第m个氨基酸的编号为n的理化特性,从而将氨基酸序列转换为(n
×
lag)维度的向量。
[0071]
具体的,如图3所示,构成人体内蛋白质的氨基酸有20种,按照侧链r基的性质可以分为5大类:带正电荷r基的氨基酸、带负电荷r基的氨基酸、极性不带电荷r基的氨基酸、侧链含非极性疏水性r基的氨基酸,以及三个特殊类别的氨基酸。如图4所示,氨基酸具有6大理化特性,其中,h,hydrophobicity,疏水性;vsc,volume of side chains,侧链的体积;p1,polarity,极性;p2,polarizability,极化性;sasa,solvent accessible surface area,溶剂可及表面积;ncisc,net charge index of side chains,侧链净电荷指数。
[0072]
利用使用上述公式将蛋白质的氨基酸序列信息表示为一定维度的向量。本工作中n=6,lag=30,这样,在上述公式计算ac
lag,n
时,n取值为1-6,lag取值为1-30,即n取1时,lag取值为1-30,n取2时,lag取值为1-30,依次类推,从而得到维度为6*30的第三特征向量。
[0073]
在一些实施例中,还包括:
[0074]
利用连接层、线性层、h个transformer模块以及归一化层构建预测模型;
[0075]
构建训练集,并对所述训练集中的每一个样本进行步骤s2-s5的处理以得到每一个样本对应的最终的特征向量;
[0076]
利用每一个样本对应的最终的特征向量对所述预测模型进行训练。
[0077]
具体的,可以选择ppi数据集,进行数据特征抽取,抽取一维序列和三维结构信息。
[0078]
ppi数据集:pan’s ppi dataset【pan,x.-y.,zhang,y.-n.&shen,h.-b.large-scale prediction of human protein
–
protein interactions from amino acid sequence based on latent topic features.j.proteome res.9,4992
–
5001(2010).】
[0079]
正样本来源于2007年6月版本的hprd数据集(human protein references database),该数据集包含38788个实验验证的蛋白质-蛋白质相互作用对,from 9630different human proteins.去除自相互作用和重复数据,最终有36630个正样本。负样本包含36480个无相互作用的蛋白质-蛋白质对。考虑到并不是所有序列在pdb数据集中都有对应的蛋白质三维结构数据,最终数据集包含25493个蛋白质-蛋白质对,其中正样本有18025个,负样本数目为7468个。
[0080]
蛋白质三维结构数据集:蛋白质三维结构信息从rcsb protein data bank(pdb,http://www.rcsb.org/pdb/)网站上提供的.pdb文件中获取,原始文件中包含了蛋白质中的原子和对应的三维坐标。
[0081]
因为这是一个二分类为题,所以模型的输出可以归为以下四类:
[0082]
●
fn:false negative,被判定为负样本,但事实上是正样本。
[0083]
●
fp:false positive,被判定为正样本,但事实上是负样本。
[0084]
●
tn:true negative,被判定为负样本,事实上也是负样本。
[0085]
●
tp:true positive,被判定为正样本,事实上也是证样本。
[0086]
因此可以用正确率、精度、召回率等用作模型性能的评估标准。
[0087]
accuracy=(tp+tn)/(p+n):正确率,即在检索结果中真正正确和真正错误的个数除以所有的样本数;
[0088]
sensitive=tp/p,灵敏度,表示的是所有正例中被分对的比例,衡量了分类器对正例的识别能力;
[0089]
specificity=tn/n,特效度,表示的是所有负例中被分对的比例,衡量了分类器对负例的识别能力;
[0090]
precesion=tp/(tp+fp):精度,即在检索后返回的结果中,真正正确的个数占整个结果的比例;
[0091]
recall=tp/(tp+fn):召回率,即在检索结果中真正正确的个数占整个数据集(检索到的和未检索到的)中真正正确个数的比例。
[0092]
如图5所示,在进行数据特征提取时,由于预测模型使用一维序列和三维结构信息作为输入,因此特征提取包括两大步:三维结构信息通过volumetric representation生成三个l*l*l的向量表示,一维氨基酸序列信息通过自协方差方法生成一个6*30的向量表示,二者都通过预训练的resnet50进行特征提取,最终得到长度为2048特征。
[0093]
如图6所示,本发明构建了一个基于自注意力机制的transformer预测模型,将上一步中得到的序列信息特征和三维结构特征作为模型的输入,现将二者concat到一起,再经过一个线性层和h个transformer模块,最后通过一个sigmoid层预测ppi的标签,如果数值大于0.5,则标签为正(意味着蛋白质和蛋白质之间有相互作用),如果数值小于0.5,则标签为负(即蛋白质和蛋白质之间没有相互作用)。
[0094]
本发明提出的方案可以同时利用一维序列信息和三维结构信息进行蛋白质相互作用的预测,利用了更多维度的信息,可以提高ppi预测的准确性。
[0095]
基于同一发明构思,根据本发明的另一个方面,本发明的实施例还提供了一种蛋白质相互作用的预测系统400,如图7所示,包括:
[0096]
获取模块401,配置为获取待预测的蛋白质的三维结构和氨基酸序列;
[0097]
缩放模块402,配置为将所述蛋白质的三维结构放置在体素为l*l*l的三维空间中,其中,所述蛋白质的三维结构的质心与所述三维空间的原点重合,并将所述蛋白质的三维结构的所有坐标进行缩放;
[0098]
第一生成模块403,配置为根据所述蛋白质的三维结构是否穿过所述三维空间中的体素得到l*l*l的形状向量,其中,若所述蛋白质的三维结构穿过体素,则值为1,否则为0;并根据所述蛋白质的三维结构中每一个氨基酸在所述三维空间的位置以及疏水性和侧链净电荷指数得到l*l*l的第一特性向量和l*l*l的第二特性向量;
[0099]
第二生成模块404,配置为利用所述氨基酸序列生成第三特征向量;
[0100]
连接模块405,配置为将所述形状向量、所述第一特征向量、所述第二特征向量以及所述第三特征向量进行连接得到最终的特征向量;
[0101]
预测模块406,配置为将所述最终的特征向量输入到训练后的预测模型中进行预测以得到待预测的蛋白质的相互作用。
[0102]
在一些实施例中,第二生成模块404还配置为:
[0103][0104]
其中,p表示蛋白质的氨基酸序列,长度为nr,m是序列中第m个氨基酸的编号,n是理化特性的编号,lag是滞后的数值,p
m,n
为第m个氨基酸的编号为n的理化特性,从而将氨基酸序列转换为(n
×
lag)维度的向量。
[0105]
在一些实施例中,缩放模块402还配置为:
[0106]
利用对所述蛋白质的三维结构的所有坐标进行缩放,其中,所述r
max
为所述三维空间中最大内切球半径。
[0107]
在一些实施例中,还包括神经网络模块,配置为:
[0108]
利用连接层、线性层、h个transformer模块以及归一化层构建预测模型;
[0109]
构建训练集,并对所述训练集中的每一个样本进行步骤s2-s5的处理以得到每一个样本对应的最终的特征向量;
[0110]
利用每一个样本对应的最终的特征向量对所述预测模型进行训练。
[0111]
基于同一发明构思,根据本发明的另一个方面,如图7所示,本发明的实施例还提供了一种计算机设备501,包括:
[0112]
至少一个处理器520;以及
[0113]
存储器510,存储器510存储有可在处理器上运行的计算机程序511,处理器520执行程序时执行如上的任一种蛋白质相互作用的预测方法的步骤。
[0114]
基于同一发明构思,根据本发明的另一个方面,如图8所示,本发明的实施例还提供了一种计算机可读存储介质601,计算机可读存储介质601存储有计算机程序610,计算机程序610被处理器执行时执行如上的任一种蛋白质相互作用的预测方法的步骤。
[0115]
最后需要说明的是,本领域普通技术人员可以理解实现上述实施例方法中的全部或部分流程,可以通过计算机程序来指令相关硬件来完成,程序可存储于一计算机可读取存储介质中,该程序在执行时,可包括如上述各方法的实施例的流程。
[0116]
此外,应该明白的是,本文的计算机可读存储介质(例如,存储器)可以是易失性存储器或非易失性存储器,或者可以包括易失性存储器和非易失性存储器两者。
[0117]
本领域技术人员还将明白的是,结合这里的公开所描述的各种示例性逻辑块、模块、电路和算法步骤可以被实现为电子硬件、计算机软件或两者的组合。为了清楚地说明硬件和软件的这种可互换性,已经就各种示意性组件、方块、模块、电路和步骤的功能对其进行了一般性的描述。这种功能是被实现为软件还是被实现为硬件取决于具体应用以及施加给整个系统的设计约束。本领域技术人员可以针对每种具体应用以各种方式来实现的功能,但是这种实现决定不应被解释为导致脱离本发明实施例公开的范围。
[0118]
以上是本发明公开的示例性实施例,但是应当注意,在不背离权利要求限定的本发明实施例公开的范围的前提下,可以进行多种改变和修改。根据这里描述的公开实施例的方法权利要求的功能、步骤和/或动作不需以任何特定顺序执行。此外,尽管本发明实施例公开的元素可以以个体形式描述或要求,但除非明确限制为单数,也可以理解为多个。
[0119]
应当理解的是,在本文中使用的,除非上下文清楚地支持例外情况,单数形式“一
个”旨在也包括复数形式。还应当理解的是,在本文中使用的“和/或”是指包括一个或者一个以上相关联地列出的项目的任意和所有可能组合。
[0120]
上述本发明实施例公开实施例序号仅仅为了描述,不代表实施例的优劣。
[0121]
本领域普通技术人员可以理解实现上述实施例的全部或部分步骤可以通过硬件来完成,也可以通过程序来指令相关的硬件完成,程序可以存储于一种计算机可读存储介质中,上述提到的存储介质可以是只读存储器,磁盘或光盘等。
[0122]
所属领域的普通技术人员应当理解:以上任何实施例的讨论仅为示例性的,并非旨在暗示本发明实施例公开的范围(包括权利要求)被限于这些例子;在本发明实施例的思路下,以上实施例或者不同实施例中的技术特征之间也可以进行组合,并存在如上的本发明实施例的不同方面的许多其它变化,为了简明它们没有在细节中提供。因此,凡在本发明实施例的精神和原则之内,所做的任何省略、修改、等同替换、改进等,均应包含在本发明实施例的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1