基于稀疏学习和随机游走的miRNA-疾病预测方法
基于稀疏学习和随机游走的mirna-疾病预测方法
技术领域
1.本发明涉及机器学习技术领域,尤其涉及基于稀疏学习和随机游走的mirna-疾病预测方法。
背景技术:
2.microrna(mirna)是一种由19-22个核苷酸组成的非编码rna,人们发现mirna的失调可能会导致许多细胞行为的失调,而且许多复杂的人类疾病与之有关。特别是,mirna在一些癌症的发生和转移中起着癌基因或肿瘤抑制剂的作用,包括乳腺肿瘤、肺肿瘤、前列腺肿瘤等。
3.但使用生物学实验来识别疾病相关的mirna既昂贵又耗时;因此,迫切需要一种简单有效的计算预测模型来预测疾病相关的mirna。
4.人们提出基于随机游走的方法来预测mirna与疾病之间的关联,如xu等人提出了基于随机游动的排序算法mirank;又如chen等人构建了imcmda算法来推断疾病-mirna相关性,将疾病相似性和mirna相似性集成到归纳完成矩阵中,以获得预测分数;但这些随机游走算法的预测模型并未能充分考虑到初始概率节点信息和相似性网络的拓扑结构信息。
技术实现要素:
5.针对现有算法的不足,本发明充分考虑到重启随机游走算法中初始概率节点信息对模型性能的影响,并且利用多层次随机游走算法在捕捉相似性网络拓扑结构特征的基础上进行了潜在关联预测。
6.本发明所采用的技术方案是:基于稀疏学习和随机游走的mirna-疾病预测方法包括以下步骤:
7.步骤一、对疾病-mirna关联矩阵进行稀疏学习,将关联矩阵分为两部分:第一部分是原始关联矩阵和低秩矩阵的线性组合,看作将有噪声的数据投影到低秩空间;第二部分是从原始矩阵中分离出来大部分元素都是0的稀疏矩阵,认为是剔除噪声或者异常值;去除稀疏部分后重构得到新的疾病-mirna关联矩阵,通过重构后的关联矩阵得到初始概率矩阵;
8.由于原始疾病-mirna已知关联中有大量未被验证的关联节点并且以前的研究中随机游走算法在确定初始概率矩阵时不能考虑到所有的节点信息;
9.进一步的,具体包括:
10.s11、将疾病-mirna关联矩阵a分解为原始关联矩阵a和稀疏矩阵e的线性组合;分别用核范数和稀疏范数对a和e进行限制;构造拉格朗日函数l,求解得到低秩矩阵x
*
;
11.s12、计算得到新的mirna-疾病关联矩阵。
12.进一步的,新的mirna-疾病关联矩阵为:a
*
=a
×
x
*
,其中,a为原始关联矩阵,x
*
为低秩矩阵。
13.进一步的,初始概率矩阵的公式为:
[0014][0015]
其中,nd是疾病的个数,nm是mirna的个数,mi为mirna,疾病为dj。
[0016]
步骤二、利用重构后的关联矩阵计算疾病和mirna的高斯核相似性,根据疾病相似性和疾病高斯核相似性构建疾病集成相似性网络;根据mirna相似性和mirna高斯核相似性构建mirna集成相似性网络;将疾病集成相似性网络和mirna集成相似性网络通过重构后的关联网络整合为一张异构网;
[0017]
步骤三、为了捕捉全局网络特征并且利用网络节点之间的关联信息,进行不同目的多层次随机游走;先分别在mirna相似性网络和疾病相似性网络上进行随机游走,得到疾病和mirna的拓扑结构相似性;结合网络的拓扑结构特征,考虑到相似性网络节点之间的关联信息,再分别在疾病和mirna拓扑结构网络上进行随机游走,有效地预测疾病-mirna潜在相似性。
[0018]
进一步的,具体包括:
[0019]
首先,在疾病相似性网络和mirna相似性网络上引入重启随机游走算法,疾病拓扑结构特征rm={rm1,rm2,
…
,rmi}和mirna拓扑结构特征rd={rd1,rd2,
…
,rdj}计算如下:
[0020][0021][0022]
其中,t代表迭代步骤,β为随机游走的衰减因子,初始概率和定义如下:
[0023][0024][0025]
将rm和rd进行标准化处理:
[0026][0027][0028]
其次,同时在疾病和mirna拓扑结构特征网络上进行随机游走并整合得到稳定概率,从而预测mirna-疾病的潜在关联;
[0029]
基于相似mirna倾向于与相似疾病关联的假设,将集成后的相似性网络、疾病相似性网络和通过稀疏化学习分解并重构之后得到新的mirna-疾病的关联网络整合到一张异构网中,分别在mirna拓扑结构网络和疾病拓扑结构网络上进行重启随机游走:
[0030]
疾病网络上:
[0031]
dr
t
=(1-γ)*r
t-1
*sd+γ*y
ꢀꢀꢀꢀꢀꢀꢀꢀ
(25)
[0032]
其中,dr是在疾病网络上进行随机游走算出的得分矩阵,mr是在mirna网络上进行随机游算出的得分矩阵,sd是上文标准化后的疾病拓扑结构特征,r是取两次随机游走得分的平均;
[0033]
mirna网络上:
[0034]
mr
t
=(1-γ)*sm*r
t-1
+γ*y
ꢀꢀꢀꢀꢀꢀꢀꢀ
(26)
[0035]
根据疾病网络和mirna网络计算平均值:
[0036][0037]
其中,t代表迭代步骤,γ为随机游走的衰减因子,y为初始概率矩阵,且r0=y。
[0038]
本发明的有益效果:
[0039]
1、通过稀疏学习方法(slm)对mirna-疾病关联矩阵进行分解和重构,以获得更丰富的关联信息,同时获得重启随机游走模型的初始概率矩阵;
[0040]
2、利用疾病相似网络、mirna相似网络、mirna-疾病关联网络构建异构网络,根据疾病和mirna的拓扑结构特征,通过多层随机游走算法得到稳定概率预测疾病-mirna潜在关联;
[0041]
3、使用全局留一交叉验证(loocv)和5折交叉验证评估模型,结果显示,全局loocv的auc值为0.9368,5-fold的平均auc值为0.9335。
[0042]
4、在案例研究中,结果表明slmrwmda模型在推断mirna疾病的潜在关联方面是有效的;
[0043]
5、将slmrwmda应用于三种高风险人类癌症进行了三种案例研究以证明本模型的适应性,结果表明slmrwmda模型有助于推断疾病-mirna的潜在相关性。
附图说明
[0044]
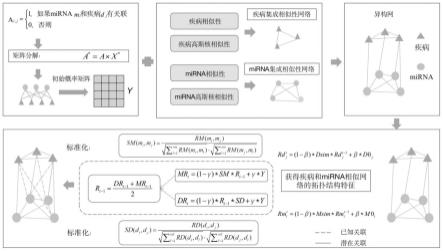
图1是本发明的基于稀疏学习和随机游走的mirna-疾病预测方法流程图;
[0045]
图2是本发明的slmrwmda与其他算法在5-fold cv中的性能对比;
[0046]
图3是本发明的slmrwmda与其他算法在全局loocv中的性能对比。
具体实施方式
[0047]
下面结合附图和实施例对本发明作进一步说明,此图为简化的示意图,仅以示意方式说明本发明的基本结构,因此其仅显示与本发明有关的构成。
[0048]
如图1所示,基于稀疏学习和随机游走的mirna-疾病预测方法包括以下步骤:
[0049]
步骤一、对疾病-mirna关联矩阵进行稀疏学习,将关联矩阵分为两部分:第一部分是原始关联矩阵和低秩矩阵的线性组合,看作将有噪声的数据投影到低秩空间;第二部分是从原始矩阵中分离出来大部分元素都是0的稀疏矩阵,认为是剔除噪声或者异常值;去除稀疏部分后重构得到新的疾病-mirna关联矩阵,通过重构后的关联矩阵得到初始概率矩阵;
[0050]
疾病-mirna关联:
[0051]
采用最新数据集版本hmdd v3.2,去除重复关系后,包含了1208个mirna和894种疾病之间的18733个关联;删除在hmdd v3.2中但不在数据库mirbase或mirtarbase中的
mirna,并删除了在hmdd v3.2中但不在mesh(美国国家医学图书馆(http://www.nlm.nih.gov)中的c类医学主题词medical subject heading)中的疾病,得到374种疾病和788种mirna之间的8968对已知关联;采取邻接矩阵来存储已知的mirna-疾病关联信息;如果疾病与mirna存在已知关联,则将其设为1,否则设为0;
[0052][0053]
进一步的,具体包括:
[0054]
首先,将疾病-mirna关联矩阵a分解为原始关联矩阵a和低秩矩阵的线性组合,包含噪声的矩阵被投影到一个信息量更大的低维空间;
[0055]
然后,添加一个稀疏矩阵,稀疏矩阵视为从原始关联矩阵中过滤出的噪声数据,其中大多数元素为0;
[0056]
最后,将上述两部分之和作为新的mirna-疾病关联矩阵;
[0057]
将关联矩阵a按式(2)分解为:
[0058]
a=a
×
x+e
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(2)
[0059]
在等式(2)中,a是低秩且稀疏,x是低秩矩阵,e是稀疏矩阵;最小化矩阵的迹范数有助于降低矩阵的秩,而稀疏范数可以识别噪声和异常值,通过矩阵分解得到a
×
x+e,然后e将它看作噪声去除;分别用核范数和稀疏范数对a和e进行限制,将公式定义为:
[0060][0061]
其中,
[0062]
||x||
*
=∑iσi(σi是矩阵的奇异值)
ꢀꢀꢀꢀꢀꢀꢀ
(4)
[0063][0064]
其中,α=0.1用于平衡低秩矩阵和稀疏矩阵的权重;因为式(3)的形式类似于鲁棒主成分分析(rpca);如果将a=a
×
x右侧中的矩阵a设为是一个单位矩阵,那么方程可以转化为:
[0065][0066]
此时,式(6)为带约束的凸优化问题;在迭代阈值(it)算法、加速近端梯度(apg)算法、精确增广拉格朗日乘子(ealm)算法和不精确增广拉格朗日乘子(ialm)算法中,将ialm算法用于将方程转换为无约束优化问题求解,以获得更精确的结果,构造拉格朗日函数l;
[0067][0068]
其中,μ(μ>0)是惩罚参数,在等式中,通过更新拉格朗日乘子y
1t
,y
2t
以及固定其他变量来最小化x,j和e,方程的解为x
*
和e
*
;如果a
ij
表示mirna的mi和疾病dj间的关联情况,那么为mirna的相似性矩阵;分解后得到x
*
和e
*
,我们去掉了噪声矩阵e
*
,通过原始的关联矩阵a和低秩矩阵x
*
的线性组合构造出新的疾病-mirna关联矩阵a
*
(a
*
=a
×
x
*
);
[0069]
通过稀疏化学习后得到的关联信息更加丰富,将新的关联矩阵中表示mirna-疾病有关联的节点作为种子节点,使得在原本稀疏初始概率矩阵变得丰富,基于重构后的关联矩阵a定以了初始概率矩阵y:
[0070][0071]
步骤二、利用重构后的关联矩阵计算疾病和mirna的高斯核相似性,根据疾病相似性和疾病高斯核相似性构建疾病集成相似性网络;根据mirna相似性和mirna高斯核相似性构建mirna集成相似性网络;将疾病集成相似性网络和mirna集成相似性网络通过重构后的关联网络整合为一张异构网。
[0072]
疾病相似性:
[0073]
将有向无环图(dag)用于表示不同疾病之间的关系,根据mesh(医学主题词表)构造出疾病的dag,获取疾病的语义信息;
[0074]
给定的疾病d表示为dag=(d,t(d),e(d)),其中,t(d)表示疾病d的所有祖先节点以及自身,e(d)表示连接父节点和子节点的边的集合,因此,疾病d对疾病d语义贡献度dd(d)定义为:
[0075][0076]
其中,δ=0.5是语义贡献衰减因子;
[0077]
进一步将疾病d的语义值定义为:
[0078]
dv(d)=∑
d∈t(d)dd
(d)
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(10)
[0079]
基于两种疾病共享dag的部分越多,其语义相似程度越大这一假设,将疾病di与疾病dj之间的语义相似性ds(di,dj)定义为:
[0080][0081]
其中,t属于两种疾病di和dj共享的dag的交集;
[0082]
mirna相似性:
[0083]
采用三种不同的标准来评估mirna的相似性;
[0084]
1、mirna序列相似性
[0085]
mirna序列来自数据库mirbase,对于每个mirna,整个成熟序列约为22个核苷酸“aucg”;基于序列信息,使用package biostrings中的成对序列比对函数“pairwise alignment”来计算相似性分数;在该函数中,差距开放惩罚设置为5,差距扩展惩罚设置为2,竞争分数设置为1,不匹配分数设置为-1;在得到序列相似性得分后,使用最大-最小归一化方法将其归一化到范围[0,1],得到的mirna序列相似性矩阵为
[0086]
2、mirna功能相似性
[0087]
基于功能相似的mirna更有可能与同一疾病相关的假设,mirna功能相似性通过其相关疾病dag的相似性来衡量;mda来自hmdd,疾病dag根据mesh描述符构建;得到表示为mirna功能相似矩阵
[0088]
3、mirna语义相似性
[0089]
mirna语义相似性由mirna靶基因和基因相关基因本体(go)注释描述;mirna靶基因信息来自mirtarbase。对于每一对mirna,维护目标基因列表,计算两个相应基因组之间的语义相似性;得到了mirna语义相似矩阵为了mirna语义相似矩阵为
[0090]
疾病和mirna的高斯相互作用谱核相似:
[0091]
基于相似的mirna通常与相似的疾病相关的假设,用基于邻接矩阵的高斯核函数计算疾病与mirna对之间的高斯相互作用谱核相似性;定义向量ip(di)表示疾病的相互作用谱,向量ip(mj)表示mirna的相互作用谱;疾病对和mirna对的高斯相互作用谱核相似性矩阵gd、gm计算如下:
[0092]
gd(di,dj)=exp(-θd||ip(di)-ip(dj)||2)
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(12)
[0093]
gm(mi,mj)=exp(-θm||ip(mi)-ip(mj)||2)
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(13)
[0094]
其中,调节系数θd和θm计算如下:
[0095][0096][0097]
其中,θ
′d和θ
′m是原始带宽,值设为1,nd是疾病的个数,nm是mirna的个数。
[0098]
疾病和mirna的集成相似性:
[0099]
对于疾病,构建集成相似性矩阵dsim整合疾病的语义相似性和高斯相互作用谱核相似性:
[0100][0101]
对于mirna,构建集成相似性矩阵msim整合mirna的相似性(见式18)和高斯相互作用谱核相似性:
[0102][0103][0104]
步骤三、为了捕捉全局网络特征并且利用网络节点之间的关联信息,进行不同目的多层次随机游走;先分别在mirna相似性网络和疾病相似性网络上进行随机游走,得到疾病和mirna的拓扑结构相似性;结合网络的拓扑结构特征,充分考虑到相似性网络节点之间的关联信息,再分别在疾病和mirna拓扑结构网络上进行随机游走,更能有效地预测疾病-mirna潜在相似性;
[0105]
进一步的,具体包括:
[0106]
不同于传统的双随机游走,在由疾病相似网络与mirna相似网络通过疾病-mirna
关联矩阵所组成的异构网中进行了两种不同目的的随机游走;
[0107]
首先,在mirna相似性网络和疾病相似性网络上引入重启随机游走算法,得到疾病和mirna的网络拓扑特征矩阵,再将其标准化;
[0108]
进一步的,具体包括:
[0109]
首先,在疾病相似性网络和mirna相似性网络上引入重启随机游走算法,通过重启随机游走算法能够充分捕捉全局网络特征且能充分利用网络节点之间的关联信息;疾病拓扑结构特征rm={rm1,rm2,
…
,rm
788
}和mirna拓扑结构特征rd={rd1,rd2,
…
,rd
374
}计算如下:
[0110][0111][0112]
其中,t代表迭代步骤,β(0<β<1)为随机游走的衰减因子,初始概率和定义如下:
[0113][0114][0115]
将rm和rd进行标准化处理:
[0116][0117][0118]
接着,同时在疾病和mirna拓扑结构特征网络上进行随机游走并整合得到稳定概率,从而预测mirna-疾病的潜在关联;
[0119]
基于相似mirna倾向于与相似疾病关联的假设,将集成后的相似性网络、疾病相似性网络和通过稀疏化学习分解并重构之后得到的新mirna-疾病的关联网络整合到一张异构网中,分别在mirna拓扑结构网络和疾病拓扑结构网络上进行重启随机游走;
[0120]
疾病网络上:
[0121]
dr
t
=(1-γ)*r
t-1
*sd+γ*y
ꢀꢀꢀꢀꢀꢀꢀꢀ
(25)
[0122]
其中,dr是在疾病网络上进行随机游走算出的得分矩阵,mr是在mirna网络上进行随机游算出的得分矩阵,sd是上文标准化后的疾病拓扑结构特征,r是取两次随机游走得分的平均;
[0123]
mirna网络上:
[0124]
mr
t
=(1-γ)*sm*r
t-1
+γ*y
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(26)
[0125]
取其平均:
[0126][0127]
其中,t代表迭代步骤,γ(0<γ<1)为随机游走的衰减因子,y为初始概率矩阵,且r0=y。
[0128]
实验结果:
[0129]
使用两种交叉验证来评估本发明的slmrwmda模型的性能,即5折交叉验证和全局留一交叉验证;将slmrwmda模型与其他五种现有技术方法如:imcmda、mdhgi、brwh、mclpmd、lplns进行比较;在五折交叉验证中,首先将所有已知的mirna-疾病关联随机分为5个大小相同的子集。每个子集依次作为测试样本,剩余四个子集作为训练样本。然后将测试集中每个测试样本的分数与所有候选分数样本进行比较,最后,获取测试样本的得分排名。而在全局留一验证中,我们将每对已知的疾病-mirna关联依次用作测试样本,而其余的mirna-疾病则作为训练样本;同样,最后算出得分排名。最终,通过计算受试者工作特征(roc)曲线和计算roc曲线下面积(auc)来评估预测性能(如图2、3);表1为5折交叉验证和全局留一交叉验证的auc结果,可以看出,本方法的性能最好;其中,全局留一的auc为0.9368,5折交叉验证的auc为0.9350;
[0130]
此外,为了验证本发明的重构关联矩阵以确定初始概率矩阵方法的有效性,进行了消融实验。在不考虑初始概率矩阵的情况下得到的5折交叉验证的auc为0.9228,由此看出,本发明的slmrwmda模型对初始概率节点的考虑是有效的。
[0131]
表1 slmrwmda与其他五种算法的对比
[0132]
模型名称5折交叉验证全局留一交叉验证imcmda0.76870.7767mdhgi0.87830.8955brwh0.85890.8629mclpmda0.92910.9305lplns0.91830.9178本模型0.93500.9368
[0133]
越来越多的证据表明,许多mirna在癌症的发生和发展中起着重要作用,某些mirna的表达水平与肿瘤的发生、复发、转移等密切相关;因此,为了验证mdbrwrmda预测未知疾病的关联能力,进行三种案例研究来进行验证;第一类案例研究利用hmddv3.2中已知疾病-mirna关联作为训练数据集;通过slmrwmda模型对人类重要疾病(食管癌)潜在相关mirna进行打分,根据得分对每个候选mirna进行排序,最后,使用dbdemc和mir2disease疾病-mirna关联数据库对预测列表中的前50名mirna进行验证。
[0134]
通过对食管癌进行上述第一类案例,结果显示,前10名中的9个mirna,前20名mirna中的19个mirna和前50名中的41个mirna被数据库dbdemc(简记为
①
)和mir2disease(简记为
②
)数据库中至少有一个被验证,结果如表2所示。
[0135]
表2预测出的前50名食管癌相关mirna在数据库中的验证
[0136][0137][0138]
为了证明mdbrwrmda模型对新疾病(即没有已知相关mirna的疾病)的预测能力,选择肺癌进行第二种案例研究;通过将训练数据中的与肺癌已知相关的mirna重置为未知,并将肺癌作为新的疾病。同样,通过mdbrwrmda模型对肺癌潜在相关的mirna进行打分,根据得分对每个候选mirna进行排序,最后使用dbdemc、mir2disease以及hmddv3.2数据库对预测列表中的前50名mirna进行验证。结果显示(表3所示,其中表格的第一列和第三列分别记录预测出的前1-25和26-50个mirna),前10个和前20个中的所有和前50个中的49个mirna被数据库dbdemc(简记为
①
)、mir2disease(简记为
②
)和hmdd3.2(简记为
③
)数据库中至少有一个被验证。
[0139]
表3预测出的前50名肺癌相关mirna在数据库中的验证
[0140]
[0141][0142]
第三类案例研究基于hmdd v2.0中的数据,通过mdbrwrmda模型对人类乳腺癌潜在相关mirna进行打分,根据得分对每个候选mirna进行排序,最后,使用dbdemc、mir2disease以及hmddv2.0疾病-mirna关联数据库对预测列表中的前50名mirna进行验证。
[0143]
结果表明,前10名和前20名中的所有以及前50名中的47个预测的mirna都被数据库dbdemc(简记为
①
)、mir2disease(简记为
②
)和hmdd v2.0(简记为
③
)中的至少一个验证结果如下表4所示:
[0144]
表4预测出的前50名乳腺癌相关mirna在数据库中的验证
[0145]
[0146][0147]
本发明使用全局留一交叉验证(loocv)和5折交叉验证评估模型,结果显示,全局loocv的auc值为0.9368,5-fold的平均auc值为0.9335。
[0148]
在案例研究中,结果表明slmrwmda在推断mirna疾病的潜在关联方面是有效的。
[0149]
将slmrwmda应用于三种高风险人类癌症进行了三种案例研究以证明本模型的适应性,结果表明slmrwmda模型有助于推断疾病-mirna的潜在相关性。
[0150]
以上述依据本发明的理想实施例为启示,通过上述的说明内容,相关工作人员完全可以在不偏离本项发明技术思想的范围内,进行多样的变更以及修改。本项发明的技术性范围并不局限于说明书上的内容,必须要根据权利要求范围来确定其技术性范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1