一种基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型及其构建方法
1.本发明属于生物医药技术领域,具体涉及一种基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型及其构建方法。
背景技术:
2.免疫疗法使黑色素瘤、非小细胞肺癌(nsclc)、乳腺癌和膀胱癌等癌症的治疗发生了革命性的变化。程序性死亡配体-1(pd-l1)表达和组织肿瘤突变(tmb)等生物标志物可作为免疫治疗疗效的预测指标。目前,普遍以1%的pd-l1表达量作为临界值将患者分为pd-l1阳性(≥1%)患者和pd-l1阴性(<1%)患者。在包括keynote-052、checkmate024等多个研究中显示,免疫治疗临床疗效在pd-l1阳性中优于pd-l1阴性患者。一些研究发现,放射学反应与pd-l1表达之间没有很强的相关性,高tmb的界值在不同类型的肿瘤中有所不同。目前迫切需要制定适当的预测策略,以帮助确定哪些潜在人群可以从免疫治疗中受益,并提供临床决策指南。同时,免疫细胞上pd-l1表达更能普遍反映由γ干扰素(ifn-γ)诱导的适应性调节,伴随着肿瘤浸润淋巴细胞和效应t细胞的增加。而肿瘤细胞上pd-l1表达反映了pd-l1基因的表观遗传失调,与免疫浸润不良、硬化/纤维增生基质和间充质分子特征所描述的独特组织学相关。肿瘤细胞或免疫细胞上的pd-l1表达可以独立地减弱抗癌免疫力,并强调免疫细胞在调节抗肿瘤t细胞应答中的功能重要性。然而,仍有一部分患者不能从免疫治疗中获益。
3.如上所述,pd-l1表达与tmb被认为是较为可靠的疗效预测生物标志物。但是,有研究发现尽管pd-l1表达作为抗pd-1/pd-l1免疫治疗相关标志物具有生物学意义,但仍有相当部分pd-l1表达阴性的肿瘤患者对pd-l1抑制剂有临床疗效,限制了部分患者潜在的生存获益。故不能仅以pd-l1表达为指导来区分最佳受益患者。同时,pd-l1免疫组织化学检测作为抗pd-1/pd-l1治疗疗效预测标志物可靠性差可能是多变量的结果。首先,pd-l1表达受多种机制调节,包括mapk和pi3k或akt途径,转录因子hif1、stat3和nfkb以及表观遗传因子。也可以由肿瘤微环境中其他免疫细胞表达。pd-l1表达可以是暂时性的,并且可以存在患者之间甚至肿瘤内pd-l1表达异质性。因此,在一个时间点或仅在一个肿瘤部位或一个肿瘤的一部分进行肿瘤采样,可能不能准确反映患者pd-1或pd-l1的状态。其次,pd-l1有免疫组织化学抗体、检查技术、检测环境和不同的pd-l1阳性阈值。例如,22c3抗pd-l1抗体克隆用于评估pembrolizumab研究中的pd-l1表达,而抗体28-8克隆用于nivolumab研究。这些研究中pd-l1表达阳性阈值各不相同,其中一些使用1%或更多阈值,另一些使用50%或更多阈值。然而,没有研究报道阳性预测值或阴性预测值接近100%。
4.针对tmb指标,有研究发现一些具有高负荷体细胞突变(tmb-high)的肿瘤对免疫检查点抑制剂无反应。因此,tmb能否成为预测免疫治疗疗效的关键很可能在于突变的质量而不是数量。除了总突变和新抗原肿瘤负荷之外,新抗原肿瘤内异质性低对免疫疗法反应也非常重要。
技术实现要素:
5.针对上述问题,本发明的目的在于提供一种基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型及其构建方法,该肿瘤免疫治疗疗效预测模型具有更高的预测能力。
6.为实现上述目的,本发明采取的技术方案为:一种用于肿瘤免疫治疗疗效预测的免疫基因评分,所述免疫基因评分的公式为:immune gene score=0.4189*pc1-0.4501*pc2,其中pc1为cox系数为正的免疫基因的表达,pc2为cox系数为负的基因的表达。
7.肿瘤免疫微环境(tme)是一种多层面的细胞环境,它不仅能限制肿瘤的发展,还能影响抗肿瘤治疗反应和肿瘤进展。研究表明,肿瘤细胞可以激活不同的免疫通路,产生免疫抑制条件,转化tme。全面了解tme是准确预测临床疗效的突破口。tme内的免疫细胞也对肿瘤的发生起决定作用。有研究表明,人白细胞抗原(hlas)可影响tme的免疫细胞浸润和基质细胞反应,失去通过hlas提交新抗原的能力可能促进免疫侵袭,促进肿瘤进展。本技术发明人通过大量实验研究,对患者的转录组rna测序(rna-seq)数据进行了个体患者水平的分析,选取关键免疫细胞、hlas和免疫检查点,结合tme这三个方面的免疫相关基因构建免疫基因评分,通过免疫基因评分公式能够将患者分为高免疫基因评分组和低免疫基因评分组,即通过免疫基因评分可以区分肿瘤免疫治疗患者的总生存期。
8.本发明还提供所述的用于肿瘤免疫治疗疗效预测的免疫基因评分的构建方法,包括以下步骤:
9.(1)收集患者的rna-seq数据,分为训练队列和验证队列;
10.(2)通过lasso算法对训练队列的rna-seq数据进行分析,筛选出免疫细胞、人类白细胞抗原和免疫检查点三个层面的免疫基因;
11.(3)对免疫细胞、人类白细胞抗原和免疫检查点三个层面的免疫基因进行整合,采用无监督聚类进行差异分析,并通过主成分分析算法构建得到免疫基因评分;
12.(4)在验证队列中对免疫基因评分公式进行验证。
13.本发明的免疫基因评分的构建方法中,采用差异分析具体为:取免疫细胞、人类白细胞抗原和免疫检查点三个层面的免疫基因中表达上调的基因取并集作为a类基因,取免疫细胞、人类白细胞抗原和免疫检查点三个层面的免疫基因中表达下调的基因取并集作为b类基因,并去除a类基因和b类基因中的并集。其中主成分分析算法具体为:对a类基因进行主成分分析得到对a类基因信息解释最大的一个主成分,即为pc1,对b类基因进行主成分分析得到对b类基因信息解释最大的一个主成分,即pc2。
14.本发明还提供所述的免疫基因评分在制备用于预测肿瘤免疫治疗疗效的产品中的应用。
15.本发明还提供所述的免疫基因评分在构建基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型中的应用。
16.本发明还提供免疫基因评分、tmb评分和lncrna评分在联合构建肿瘤免疫治疗疗效预测模型中的应用。
17.本发明还提供一种基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型,采用免疫基因评分、tmb评分和lncrna评分联合构建得到。
18.作为本发明所述的基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型的优选实施方式,所述预测模型为:tme signature=-0.67729*immune_gene_score+lncrnascore*
0.93203-0.02610*tmb。
19.本发明的预测模型中的lncrnascore=
–
(dlgap1-as1*0.0388404669)
–
(ramp2-as1*0.1155662963)-(bves-as1*0.0553720312)-(lipe-as1*0.1033882346)+(linc01118*0.0796557151)
–
(fgd5-as1*0.0289597115)-(al513365.2*0.20586808)-(emc1-as1*0.0694844742)+(ftx*0.0852566063)+(snhg15*0.1004314629)
–
(tmem147-as1*0.0720185014)+(ttn-as1*0.0540188921)+(flg-as1*0.0526396975)-(cdkn2b-as1*0.0064524961)+(usp2-as1*0.0747066713)
–
(sbf2-as1*0.1293691962)-(ac113382.1*0.2179618875)+(nop14-as1*0.0644565543)+(ac012636.1*0.1939511817)-(linc01605*0.0168478032)+(pcat1*0.1453946597)+(brwd1-as2*0.0298250867)-(usp3-as1*0.0691270103)+(vps9d1-as1*0.0003103997)-(ac093249.6*0.0139283015)
–
(snhg8*0.0014401603)-(fut8-as1*0.0852427371)+(nkila*0.1318600645)-(slfnl1-as1*0.0472823654);所述tmb通过采取肿瘤组织样本检测得到。
20.作为本发明所述的基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型的优选实施方式,所述预测模型的阈值通过survimer的surv_cutpoint函数确定。
21.本发明还提供所述的肿瘤免疫治疗疗效预测模型在制备用于肿瘤免疫治疗疗效预测的产品中的应用。
22.本发明的有益效果为:本发明提供了一种基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型,本发明选取关键免疫细胞、hlas和免疫检查点,结合tme这三个方面的免疫相关基因构建免疫基因评分,并通过联合免疫基因评分、tmb评分和lncrna评分构建基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型,该预测模型相较于免疫基因评分、tmb评分、lncrna评分等单因素预测策略,能够显著提高肿瘤免疫治疗疗效预测的效能,具有更高的准确性和敏感性,为肿瘤患者的治疗策略提供更准确的指导。
附图说明
23.图1为实施例1的6048例肿瘤患者具体数据图。
24.图2为实施例1中348例接受pd-l1抑制剂atezolizumab治疗的膀胱癌患者的rna-seq数据分析图。
25.图3为关键免疫细胞的选择和功能富集,其中a和b为在348例接受pd-l1形式imvigor210治疗的膀胱癌患者的rna-seq数据中,采用lasso算法筛选出5个免疫细胞;c为无监督层次聚类方法的最优聚类数为2类;d为火山图显示两个免疫细胞相关簇的差异表达基因;e为两个免疫细胞相关簇的基因本体(go)分析;f为京都基因和基因组百科全书(kegg)对两个免疫细胞相关集群的分析。
26.图4为不同肿瘤免疫微环境簇中免疫治疗的总生存获益,其中a为两种免疫细胞相关簇的免疫治疗总生存率;b为两个免疫细胞相关簇的基因热图;c为两种基于白细胞抗原的群集免疫治疗的总生存率;d为两种人类白细胞抗原集群的基因热图;e为两个免疫检查点相关聚集群的免疫治疗总生存率;f为两个免疫检查点相关集群的基因热图。
27.图5为关键人类白细胞抗原的选择与功能富集,其中a和b为在348例接受pd-l1形式imvigor210治疗的膀胱癌患者的rna-seq数据中,采用lasso算法筛选出7种人白细胞抗原(hlas);c为无监督层次聚类方法的最优聚类数为2类;d为火山图显示了两个hla相关簇
的差异表达基因;e为两个hla相关聚类的基因本体(go)分析。
28.图6为关键免疫检查点的选择和功能的富集,其中a和b为在348例接受pd-l1形式imvigor210治疗的膀胱癌患者的rna-seq数据中,采用lasso算法选择4个免疫检查点;c为无监督层次聚类方法的最优聚类数为2类;d为两个免疫检查点相关簇差异表达基因的火山图;e为两个免疫检查点相关簇的go分析;f为对两个免疫检查点相关簇的kegg分析。
29.图7为三个免疫相关基因簇及其功能情况,其中,a为三个免疫基因相关簇的基因热图;b和c为在imvigor210试验和validation-4中,免疫治疗在这些集群中的总生存受益有显著差异;d和e为缺氧评分和代谢功能注释有显著差异。
30.图8为采用无监督层次聚类方法将免疫相关基因划分为3个免疫基因相关簇。
31.图9为免疫基因相关簇不同基因组功能,其中a-d为三个免疫基因相关簇的不同共表达基因模块和不同的基因表达模式;e-g为3个免疫基因相关簇的go分析;h为三个免疫基因相关簇的kegg分析。
32.图10为两组免疫基因评分组具有不同的基因表达和基因组功能,其中,a为高免疫基因评分组的oncoprint;b为低免疫基因评分组的oncoprint;c为差异表达基因火山图;d为两组免疫基因评分的go分析;f为两组免疫基因评分的kegg分析。
33.图11为两个免疫基因评分组可以很好地区分患者的生存获益,其中a为348例经pd-l1形式imvigor210治疗的膀胱癌患者的总生存期;b为97例免疫治疗gse78220和tcga-skcm的黑色素瘤患者的总生存期;c为97例经gse78220和tcga-skcm免疫治疗的黑色素瘤患者的无进展生存期;d为来自gse135222的27例接受免疫治疗的非小细胞肺癌患者的总生存率;e为来自癌症基因组图谱(tcga)的5576例标准治疗的癌症患者的总生存率。
34.图12为两组免疫基因评分组cd274和cyt的表达,其中,a和e为imvigor210治疗348例膀胱癌患者cd274和cyt表达的差异;b和f为97例免疫治疗gse78220和tcga-skcm的黑色素瘤患者cd274和cyt表达的差异;c和g为gse135222免疫治疗27例非小细胞肺癌患者cd274和cyt表达的差异;d和h为来自tcga的5576例标准治疗的癌症患者cd274和cyt表达的差异。
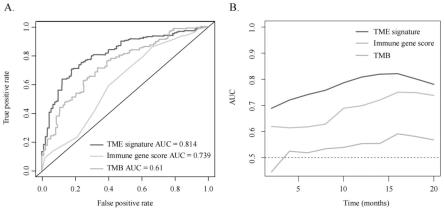
35.图13为肿瘤免疫微环境特征对肿瘤免疫治疗的生存效益有较好的预测作用,其中a为预测20个月免疫治疗疗效的免疫基因评分、tmb、肿瘤免疫微环境(tme)特征的受试者工作特征(roc)曲线;b为免疫基因评分、tmb、tme标志预测免疫治疗疗效随时间变化的roc曲线。
具体实施方式
36.为了更加简洁明了的展示本发明的技术方案、目的和优点,下面结合具体实施例和附图详细说明本发明的技术方案。
37.实施例1
38.本发明实施例提供一种用于肿瘤免疫治疗疗效预测的免疫基因评分的构建方法。
39.本实施例的采用r软件(r software,http://www.r-project.org)、pythom软件(https://www.pythom.com/)进行统计学分析。具体统计学方法如下:
40.统计分析一般原则:除特殊说明外,所有的统计检验都采用α=0.05的双侧检验,置信区间均采用双侧95%的置信区间估计。定量指标采用例数、均数、标准差、中位数、上下四分位数、最小值和最大值进行统计描述,分类指标采用各类的例数和百分比进行统计描
述等。两组基线均衡性的比较分析采用t检验、χ2检验、fisher精确检验或秩和检验等。
41.免疫相关基因的选择:采用r包方差稳定变换对计数数据进行质量控制和归一化处理。利用包含577个人免疫细胞标记基因对转录本数据进行24种人免疫细胞表型标记。采用r语言的glmnet包进行最小绝对收缩和选择算法(lasso算法)选择关键免疫细胞、人类白细胞抗原(hlas)和免疫检查点,惩罚参数优化采用5倍交叉验证。利用r语言的cancersubtypes包进行无监督层次聚类方法(k-means),在不同聚类中找出稳定性最高、模糊性最低的最优聚类数。为了筛选差异表达基因,我们使用r语言的limma包计算转录本fold-change,将大于2且p值调整后小于0.05的转录本作为截断值。通过基因热图和火山图显示差异表达基因的分布和表达水平。
42.免疫治疗疗效预测模型的构建:采用主成分分析(pca),提取相关系数为正和负的主成分各1个进行免疫基因评分计算,免疫基因评分=a*pc1-b*pc2;其中pc1为cox系数为正的免疫基因的表达,pc2为cox系数为负的基因的表达。在获得每个基因评分的预后值后,建立公式定义每个患者的免疫基因评分。然后结合免疫基因评分、tmb、lncrna评分构建预测模型,预测免疫治疗的生存。
43.内部机制途径探索:利用r语言的clusterprofiler包对基因本体(go)和京都基因和基因组百科全书(kegg)进行分析。当p值和错误发现率小于0.05时,认为kegg通路和go项的细胞成分、分子功能和生物学过程有统计学意义。通过加权基因共表达网络分析(wgcna)鉴定共表达基因模块。基因集富集分析(gene set enrichment analysis,gsea)用于定量基因功能通路的比例。通过r语言的gsva包,利用broad institute富集算法对分子特征库(msigdb)中的一个基因进行基因集变异分析(geneset variation analysis,gsva),计算缺氧评分。基因富集的代谢途径采用xiao等报道的算法进行。cyt的表达量定义为cd8a、cd8b、gzma、gzmb、prf1这5个基因的平均表达量。
44.本实施例的数据来源:将6048例肿瘤患者数据分为训练队列和三个验证队列,其中472例患者接受免疫治疗,5576例患者接受标准治疗(治疗策略未公布),详细数据如图1所示。训练队列来自单臂、ii期、多中心的imvigor210试验的348例接受pd-l1抑制剂atezolizumab治疗的膀胱癌患者。validation-1为来自gse78220队列的69例接受抗pd-1治疗的黑色素瘤患者,validation-2为来自gse135222队列的27例接受抗pd-1/pd-l1治疗的非小细胞肺癌患者,validation-3为来自tcga-skcm的28例接受免疫治疗的黑色素瘤患者,validation-4来自tcga的5,576例泛肿瘤患者。这些个体患者水平的数据从geo数据库(http://www.ncbi.nlm.nih.gov/geo/)和从ucsc xena浏览器(gdc hub:https://gdc.xenahubs.net)下载的癌症基因组图谱(tcga)。rna-seq数据被转化为每千碱基百万值的转录本。
45.具体实验方法:
46.1、筛选与免疫治疗有关的肿瘤免疫微环境关键影响因素
47.通过lasso算法对imvigor210试验的348例接受pd-l1抑制剂atezolizumab治疗的膀胱癌患者的rna-seq数据分析,筛选出了5个免疫细胞、8个hlas和4个免疫检查点。所有免疫细胞、hlas和免疫检查点的详细情况见图2。
48.采用lasso算法,在从22个免疫细胞中筛选出5个关键免疫细胞,包括γδt细胞、激活的自然杀伤(nk)细胞、巨噬细胞m1、巨噬细胞m2和激活的大量细胞(如图3a和图3b所示)。
根据不同的基因表达,这些细胞被进一步分为两个不同的免疫细胞簇(如图3c和图3d所示)。这两个不同免疫细胞簇在免疫治疗中具有不同的os获益(hr,3.23;95%ci,1.85~5.67;p《0.001)(图4a),两个簇的差异表达基因由基因热图(图4b)显示。两个聚类的功能注释和通路在趋化因子信号通路以及响应干扰素-γ、细胞因子受体结合和趋化因子活性的基因组生物学过程中富集(如图3e和图3f所示)。
49.采用lasso算法,在23个hlas中筛选出7个关键hlas,包括hla-c、hla-dma、hla-doa、hla-dqb1、hla-drb3、hla-f-as1、hla-g和hla-b(如图5a和图5b所示),根据基因表达的差异将其分为两个不同hlas簇(如图5c和图5d所示)。这两个不同hlas簇在免疫治疗中具有不同的os获益(hr,1.43;95%ci,1.09-1.89;p=0.010)(图4c),通过基因的基因热图(图4d)显示了两个簇的表达基因差异。两个簇的功能注释和通路在水解酶活性、分泌颗粒膜和肽酶调节因子活性上富集(图5e)。
50.采用lasso算法,在22个免疫检查点筛选出4个关键免疫检查点,包括cd276,ido1,lag3和sirpa(如图6a和图6b所示)。根据差异基因表达将其分为两个不同免疫检查点簇(如图6c和图6d所示)。这两个不同的免疫检查点簇在免疫治疗中具有不同的os获益(hr,1.46;95%ci,1.13-1.89;p=0.004)(图4e),两个簇的差异表达基因由基因的热图(图4f)显示。这两个簇在细胞因子-细胞因子受体相互作用和趋化因子信号通路中有功能注释和通路富集,在t细胞激活中有基因组生物学过程(如图6e和图6f所示)。
51.为了结合免疫细胞、hlas和免疫检查点的差异表达基因一同进项免疫治疗获益预测,对免疫细胞、hlas和免疫检查点这三个层面的1360个免疫相关差异表达基因进行整合分析,并利用无监督聚类将其分为3个免疫基因相关簇(如图7a、图8所示)。接受免疫治疗患者的os获益在3个聚类间存在显著差异(p《0.001,图7b),这一结果在验证队列validation-4接受标准治疗的患者中得到验证,即免疫基因相关聚类可以显著区分患者的生存差异(p《0.001,图7c)。
52.在这些聚类中,计算缺氧评分并进行代谢功能注释,结果显示三个免疫基因相关聚类存在显著的缺氧和代谢状态差异(p《0.001,图7d和图7e),其中缺氧评分最高的聚类a上调了糖链生物合成和代谢的代谢功能;低缺氧评分的聚类c伴脂质代谢功能上调。此外,在三个聚类中发现了不同的共表达基因模块和不同的基因表达模式(图9a-d),显示了不同的基因组功能,聚类a表达于rap1信号通路,而聚类c表达于mapk信号通路(图9e-h)。
53.利用训练队列的免疫相关基因数据集,采用pca算法构建预测免疫治疗疗效的免疫基因评分。患者免疫基因评分计算公式:免疫基因评分=0.4189*pc1-0.4501*pc2。其中pc1为cox系数为正的免疫基因的表达,pc2为cox系数为负的基因的表达。将患者分为高免疫基因评分组和低免疫基因评分组,最佳阈值为0.22。两组存在基因表达差异(图10a和图10b)。通过筛选免疫相关基因与患者免疫治疗生存受益的相关性,阳性基因107个,阴性基因71个(图10c),两组差异表达基因涉及补体与凝血级联、细胞因子-细胞因子受体相互作用、系统性红斑狼疮、病毒蛋白与细胞因子和细胞因子受体相互作用、go富集分析发现高组和低组在dna复制、细胞-底物连接、染色体区域、细胞器裂变等方面具有不同的基因组功能(图10e)。kegg通路富集分析发现两组在pi3k-akt信号通路、黏附灶、细胞周期、dna复制等方面表达不同(图10f)。
54.针对患者的os获益,高免疫基因评分组患者的os显著高于低免疫基因评分组(hr,
2.413;95%ci,1.76-3.307;p《0.0001,图11a)。在多个队列中进行验证分析,包括采用免疫治疗的黑色素瘤患者(validation-1和validation-3)(os:hr=3.71;95%ci,1.13-12.18,p=0.021,无职业性生存:hr=1.949;95%ci,1.121~3.387;p=0.018,图11b和图11c),接受免疫治疗的nsclc患者形成validation-2(os:hr,3.36;95%ci,1.26~8.94;p=0.015;图11d)。此外,免疫基因评分可区分validation-4标准治疗的患者总生存期(图11e)。
55.此外,高免疫基因评分组比低免疫基因评分组cd274(pd-l1)和cyt表达高(图12a,图12e)。这些发现也在多队列中得到验证,包括免疫治疗形式validation-1和validation-3的黑色素瘤患者(图12b,图12c,图12f,图12g),以及来自validation-4的标准治疗的癌症患者(图12d,12h)。
56.实施例2
57.本发明实施例提供一种基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型,该模型采用实施例1的免疫基因评分、tmb评分、lncrna评分和lncrna评分联合构建得到。
58.分别采用本实施例的基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型、tmb评分和免疫基因评分进行肿瘤免疫治疗疗效预测,预测结果如图13所示。由图8的受试者工作特征(roc)曲线可知,tme评分的auc值为0.814、tmb评分的auc值为0.61、lncrna评分的auc值为0.791、免疫基因评分的auc值为0.737,由此说明,与免疫基因评分、tmb评分、lncrna评分等单因素预测策略相比,本实施例的基于肿瘤免疫微环境的肿瘤免疫治疗疗效预测模型显著提高了免疫治疗疗效预测的准确性。
59.最后应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1