一种单分子水平的SMRT-seq质量控制方法
一种单分子水平的smrt-seq质量控制方法
技术领域
1.本发明涉及一种甲基化检测技术领域,尤其涉及一种单分子水平的smrt-seq质量控制方法。
背景技术:
2.表观遗传学是指生物体非基因序列变化而引起基因的功能和表达水平发生可遗传改变的研究,其与肿瘤发生、精神疾病和免疫组学代谢等生物学过程息息相关。
3.生物体内的表观遗传现象主要包括dna甲基化修饰、染色体重塑、组蛋白修饰和非编码rna调控等。dna甲基化是最早被发现的表观遗传修饰之一,参与许多重要的细胞进程,如基因组印记、x染色体灭活、转录抑制和胚胎发育等。由于dna的双螺旋结构,dna上核苷酸的甲基化不仅影响染色质的开放性,也通过改变核苷酸分子构象影响dna的三维结构,改变其与蛋白质的结合的方式,从而影响基因表达。根据中心法则,dna甲基化修饰是环境影响生物体代谢调控和疾病发生的根本途径。目前主要研究的dna甲基化有6ma、5mc和4mc,其中6ma在细菌界非常普遍,也存在于低等真核生物中,但随着近几年测序技术的发展,6ma被证明存在于多种高等真核生物中,且与核小体定位、神经发育、癌症发生等过程高度相关。
4.n6-甲基脱氧腺苷(6ma)和c5-甲基胞嘧啶(5mc)是生物体内常见的dna甲基化修饰,在真核和原核生物生命过程中都具有至关重要的作用。真核生物中最常见的dna修饰是c5-甲基胞嘧啶(5mc),目前bs-seq(亚硫酸氢盐测序)是5mc甲基化测序的主要检测方法,需对dna样品进行亚硫酸盐处理,使非甲基化的c变成u,而甲基化的c保持不变,从而使其可以在后续的测序或者杂交结果中区分出来。得益于这项技术的广泛应用,5mc在原核和真核生物体中均得到了充分研究。而6ma因丰度普遍较低和受到检测技术限制,在识别方法上尚未取得重大的进展。
5.三代单分子测序技术给这项研究带来了曙光,测序公司将测序中荧光脉冲信号的时间跨度(inter pulse duration,ipd)与整体ipd期望值的比率(ipdratio)用于检测碱基的状态,并且在测序分析软件中加入了识别ipdratio显著差异的碱基修饰功能。然而该模块是基于原核生物进行开发,是否适用于真核生物一直存在质疑,同时真核生物检测中存在大量假阳性结果。
6.该方案在分析原始测序数据时发现测序数据分析软件在统计位点ipd时,选取了所有读长的ipd的均值,代表位点的真实ipd,这可能忽视了读长的局部特征,致使检测准确率难以提高。
技术实现要素:
7.本发明主要解决现有技术以读长ipd的均值代表点位的真实ipd,忽略读长的局部特征,检测准确率低的问题;提供一种单分子水平的smrt-seq质量控制方法。
8.本发明的上述技术问题主要是通过下述技术方案得以解决的:一种单分子水平的smrt-seq质量控制方法,包括以下步骤:
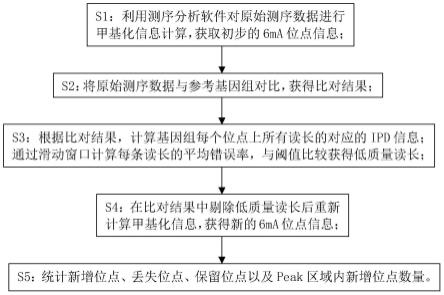
s1:利用测序分析软件对原始测序数据进行甲基化信息计算,获取初步的6ma位点信息;s2:将原始测序数据与参考基因组对比,获得比对结果;s3:根据比对结果,计算基因组每个位点上所有读长的对应的ipd信息;通过滑动窗口计算每条读长的平均错误率,与阈值比较获得低质量读长;s4:在比对结果中剔除低质量读长后重新计算甲基化信息,获得新的6ma位点信息;s5:统计新增位点、丢失位点、保留位点以及peak区域内新增位点数量。
9.本技术建立了一套纵向分布和横向比较的打分机制,通过滑动窗口统计了读长匹配在基因组上全部区域的分值,考虑读长的全部特征,剔除低质量读长,提高了检测的准确率。
10.作为优选,所述的比对结果包括含有ipd信息的比对结果和未含有ipd信息的比对结果;含有ipd信息的比对结果用于后续计算;未含有ipd信息的比对结果用于质控后构建重新计算甲基化位点的过程文件。
11.作为优选,将获得的比对结果进行正负链区分。
12.如染色体数量较多,可以自定义按照染色体拆分文件,使用parallel命令并行计算,提高计算效率。
13.作为优选,基因组每个位点上所有读长的对应的ipd信息的计算过程为:通过阈值比较,筛选长度与质量符合要求的读长;将6ma位点信息与对应读长的位置信息进行匹配。
14.作为优选,滑动窗口计算每条读长的平均错误率的过程为:将一个位点的ipd数据按大小排列,取平均值正负一个标准差范围内的ipd数据为该位点的正常值,否则为异常值;在染色体上以长度为l,步长为d的窗口滑动检索,计算每个窗口中读长中ipd异常的位点占该读长全部位点的比例。
15.作为优选,根据指定阈值找出低质量读长的id,其具体方法如下:将同一读长的所有窗口及其错误率合并,统计读长对应的所有窗口错误率的均值,记为该读长的最终得分;输出读长的最终得分低于指定阈值的低质量读长id。
16.剔除低质量读长,提高了检测的准确率。
17.本发明的有益效果是:建立了一套纵向分布和横向比较的打分机制,通过滑动窗口统计了读长匹配在基因组上全部区域的分值,考虑读长的全部特征,剔除低质量读长,提高了检测的准确率。
附图说明
18.图1是本发明的smrt-seq质量控制方法流程图。
19.图2是本发明的ipd质控模型打分机制示意图。
具体实施方式
20.下面通过实施例,并结合附图,对本发明的技术方案作进一步具体的说明。
21.实施例:本实施例的一种单分子水平的smrt-seq质量控制方法如图1所示,包括以下步骤:s1:利用测序分析软件对原始测序数据进行甲基化信息计算,获取初步的6ma位点信息。
22.在本实施例中,原始测序数据序列通过pacbio测序技术获得,使用smrtanalysis软件对原始测序数据进行计算,获取初步的6ma位点信息。
23.s2:将原始测序数据与参考基因组对比,获得比对结果。
24.比对结果包括含有ipd信息的比对结果和未含有ipd信息的比对结果。在本实施例中,使用blasr将原始pacbio测序数据序列与初步修饰后的基因组进行比对,获取含有ipd信息的比对结果以及未含有ipd信息的比对结果。
25.含有ipd信息的比对结果用于后续的错误率计算。
26.未含有ipd信息的比对结果用于质控后构建重新计算甲基化位点的过程文件。
27.未含有ipd信息的比对结果,便于质控后修改sam文件,重新构建计算6ma位点的中间文件。
28.s3:根据比对结果,计算基因组每个位点上所有读长的对应的ipd信息;通过滑动窗口计算每条读长的平均错误率,与阈值比较获得低质量读长。
29.将获得的比对结果进行正负链区分,在本实施例中,使用samtools区分正负链和自定义拆分染色体。如染色体数量较多,可以自定义按照染色体拆分文件,使用parallel命令并行计算,提高计算效率。
30.计算基因组每个位点上的所有读长对应ipd信息,为后续计算错误率做准备。
31.在本实施例中,使用python计算基因组每个位点上的所有读长对应ipd信息方法如下:根据阈值比较筛选掉长度较短和质量较低的读长,将之前步骤中得到的6ma信息和对应读长的位置信息通过读长和位置匹配起来,为后续计算错误率做准备。
32.如图2所示,建立了一套纵向分布和横向比较的打分机制,通过滑动窗口统计了读长匹配在基因组上全部区域的分值。
33.该窗口中异常值个数和窗口长度之比则为读长在该窗口的错误率。三角形代表在单点读长的ipd分布中处于异常值,圆形代表在单点读长的ipd分布中处于正常值。错误率则为横向比较时ipd异常的位点占该读长全部位点的比例。
34.通过滑动窗口计算每条读长的平均错误率,作为打分依据。
35.滑动窗口计算每条读长的平均错误率进行打分的方法如下:选取平均值正负一个标准差内的ipd数据为该位点的正常值,否则为异常值,进行统计。
36.在本实施例中,在染色体上以5000bp为窗口长度,2000bp为步长进行滑动检索,计算每个窗口中读长的错误率。
37.根据指定阈值找出低质量读长的id,默认会输出0.1-0.9倍的指定阈值所对应的读长id列表。
38.根据指定阈值找出低质量读长的id,其具体方法如下:
通过脚本将所有读长及其错误率合并起来,并且统计读长所有窗口错误率的均值,记为该读长的最终得分,即ipd质控模型中读长的错误率。
39.s4:在比对结果中剔除低质量读长后重新计算甲基化信息,获得新的6ma位点信息。
40.在比对结果中取出低质量读长后,重新计算甲基化信息,其具体方法如下:将指定阈值下提取出的读长,使用linux grep命令从原始sam文件中过滤出来,重新利用smrtanalysis软件中甲基化检测模块再次计算6ma位点信息。
41.s5:统计新增位点、丢失位点、保留位点以及peak区域内新增位点数量。
42.将重新计算后新的6ma位点信息与原始的初步的6ma位点信息进行比对,统计新增位点,丢失位点、保留位点以及peak区域内新增位点数量。
43.本实施例将执行本实施例方法的程序的封装成九个程序执行模块,程序执行模块中包含了计算中所需要的所有shell脚本、pyhton脚本和r语言脚本,便于分步骤修改调试。
44.各程序执行模块包括:(1)第一程序执行模块:为参考基因组建立索引,利用测序分析软件对原始测序数据进行计算,获取初步的6ma位点信息。
45.(2)第二程序执行模块:将原始数据比对到参考基因组,获取带有ipd信息的比对结果。
46.(3)第三程序执行模块:获取未含有ipd信息的比对结果,便于质控后修改sam文件,重新构建aligned.cmp.h5中间文件。
47.(4)第四程序执行模块:将(2)、(3)模块中所获得的比对结果进行正负链区分,如染色体数量较多,可以自定义按照染色体拆分文件,使用parallel命令并行计算,提高计算效率。
48.(5)第五程序执行模块:计算基因组每个位点上的所有读长对应ipd信息,为后续计算错误率做准备。
49.(6)第六程序执行模块:通过滑动窗口计算每条读长的平均错误率,作为打分依据。
50.(7)第七程序执行模块:根据指定阈值找出低质量读长的id,默认会输出0.1-0.9阈值所对应的读长id列表。
51.(8)第八程序执行模块:在比对结果中取出低质量读长后,重新计算甲基化信息,得到新的6ma位点。
52.(9)第九程序执行模块:将重新计算后的结果与原始结果进行比对,统计新增位点,丢失位点、保留位点以及peak区域内新增位点数量。
53.采用本实施例的方法的实验数据如下:选取了原核数据集,包含大肠杆菌e.coli、枯草芽孢杆菌b.subtilis、金黄葡萄球菌s.aureus、李斯特菌l.monocytogenes、粪肠球菌e.faecalis、肠炎沙门氏菌s.enterica等六种细菌的数据集,其中包括pacbio rsii数据、6mamedip-seq数据以及对应参考基因组文件,具体数据来源如表1所示。用b.subtilis、e.faecalis、e.coli、l.monocytogenes、s.enterica、s.aureus等六个细菌的测试数据,同样进行ipd质控并检测6ma位点,结果如表2所示。
54.表1.原核数据集物种参考基因组pacbio数据medip-seq数据e.coliecoli_pb.fastasamn10365290gse62690b.subtillsbsubtills_pb.fastasamn09475310——e.faecalisefaecalis_pb.fastasamn09475313——s.aureussaureus_pb.fastasamn09475317——s.entericasenterica_pb.fastasamn09475319——l.monocytogeneslmonocytogenes_pb.fastasamn09475315——表2.测试数据表2.测试数据结果表明,e.faecalis新增位点最多,为1527个,其新增peak内位点数也最多,为212个;s.enterica新增位点最少,为594个,新增peak内位点数为192个;若单纯以新增peak内位点数量来评价,e.faecalis表现最好,s.aureus表现最差,仅新增88个peak内位点。六种细菌的数据以0.4为阈值的实验结果表明,丢失位点占新增位点的比例比衣藻实验结果低,可见本文ipd质控模型在原核生物中表现较好,新增位点相对多,而丢失位点相对较少。
55.应理解,实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1