一种酰胺类化合物在制备防治近视的药物中的应用
1.本发明属于医药技术领域,尤其是涉及一种酰胺类化合物在制备防治近视的药物中的应用。
背景技术:
2.目前,近视已经成为全球流行的视力障碍,尤其是在亚洲,青少年近视的患病率约为80%-90%,伴随而来的高度近视患病率达到10%-20%。有研究预测,到2050年近一半的世界人口将发展为近视,其中高度近视占世界人口的10%。近视通常指在眼睛调节放松时,平行于视轴的光线经眼的屈光系统,在视网膜前形成焦点,从而使得视网膜上成像模糊。这主要由于眼球的前后轴过长,或角膜和(或)晶状体的屈光力过大造成的。通常,调节放松状态下,眼的等效球镜度小于-0.5d时为近视,小于-6.0d时为高度近视。高度近视可引发一系列的眼部病理性改变,如后巩膜葡萄肿,视网膜脱离和高度近视性脉络膜新生血管,进而导致不可逆性视力丧失。因此,探索开发近视治疗药物是亟待解决的重要问题。
3.当前治疗近视主要通过改变眼球轴向长度或者角膜、晶状体的屈光度来实现,具体手段包括光学和手术矫正视力,以及药物延缓近视进展三大类。光学矫正是指佩戴框架眼镜、角膜接触镜,以及渐进多焦点眼镜、双焦眼镜等。光学矫正并未从根本上解决屈光不正的问题,也不能阻止高度近视导致的眼部病变。手术矫正手段主要包括角膜屈光手术、巩膜屈光手术和眼内屈光手术。手术矫正视力的方式大部分效果较好,但均有局限性,且其长期风险仍有待评估。近视治疗药物拥有巨大的潜在市场,当前主要为毒蕈碱型受体(m受体)拮抗药,以阿托品为代表。研究显示,高(1%和0.5%)、中(0.1%)、低(0.01%)浓度的阿托品滴眼液均可有效延缓近视进展,屈光度和眼轴长度均得到一定程度地改善,但具体作用机制尚无定论。目前低浓度阿托品的药物研发备受关注,中、日、美多家药企正在开发低浓度硫酸阿托品眼用制剂,且均已进入ⅲ期临床研究,但如何在接近泪液ph值条件下,保持低浓度阿托品的稳定性是一大挑战。阿托品对近视具有一定的治疗效果,但会导致眼部一系列不良反应,如瞳孔散大、畏光、睫状肌麻痹和视近模糊等。此外,阿托品对组织器官的选择性不高,对近视的疗效往往需要长期使用才能显现,而长期使用的安全性和不良反应依然无法预估。因此寻找新的、更有效的、无毒副作用的防治策略仍是研究人员亟需解决的问题。
技术实现要素:
4.有鉴于此,针对现有技术的不足,本发明提出了一种酰胺类化合物在制备防治近视的药物中的应用,为防治近视提供新的药物,该化合物可以靶定非经典wnt通路中sfrp-1蛋白,是一种对sfrp-1蛋白亲和力强,且半衰期长、作用效果明显、副作用小的sfrp-1特异性抑制剂,能够有效防治近视。
5.为达到上述目的,本发明的技术方案是这样实现的:
6.本发明一方面提供了一种酰胺类化合物在制备防治近视的药物中的应用,其中所
述化合物的结构式如式(ⅰ)所示:
[0007][0008]
化合物d的英文名称:(s)-1-phenyl-n-(1-(tetrahydrofuran-2-carbonyl)indolin-6-yl)methanesulfonamide,化学式为:c
20h22
n2o4s,相对分子量为386.4660。
[0009]
进一步的,所述如式(ⅰ)所示的化合物为sfrp-1特异性抑制剂。
[0010]
进一步的,所述如式(ⅰ)所示的化合物通过与sfrp-1蛋白的活性位点进行靶向结合,拮抗sfrp-1引起的细胞内ca
2+
浓度升高,从而抑制非经典wnt/ca
2+
/camk信号通路的过度激活,达到防治近视的目的。
[0011]
进一步的,所述如式(ⅰ)所示的化合物抑制细胞内钙离子浓度的有效工作浓度为0.4~10um。
[0012]
进一步的,所述如式(ⅰ)所示的化合物抑制细胞内钙离子浓度的有效工作浓度为0.4um。
[0013]
本发明另一方面提供了一种防治近视的药物组合物,包括如式(ⅰ)所示的化合物和药学上可接受的一种或多种物质;
[0014][0015]
进一步的,所述如式(ⅰ)所示的化合物作为有效成分,抑制细胞内钙离子浓度的有效工作浓度为0.4~10um。
[0016]
进一步的,所述如式(ⅰ)所示的化合物作为有效成分,抑制细胞内钙离子浓度的有效工作浓度为0.4um。
[0017]
进一步的,所述药物组合物的剂型为气雾制剂或液体制剂。
[0018]
本发明再一方面提供了一种如式(ⅰ)所示的化合物在制备sfrp-1特异性抑制剂药物中的应用,
[0019][0020]
本发明的技术原理:近视的病因十分复杂,主要与眼轴过度延长有关。近年来有研究者提出了一种“巩膜缺氧学说”,即模糊的视觉刺激作用于视网膜,引起视网膜的信号因子改变,进而使脉络膜变薄、血流减少,导致无血管的巩膜缺氧,胞外基质重塑,最终造成近视发生。上述学说提示,脉络膜血流减少而导致的巩膜缺氧与近视的发生、发展密切相关,但是具体的信号分子、细胞靶点及其作用机制尚不明确。
[0021]
基于此,本发明首先建立豚鼠形觉剥夺性近视(fdm)和光学离焦性近视(lim)模
型,通过对眼球后极部组织进行蛋白质组学分析和蛋白印迹验证,结果显示从造模后1周起,cochlin基因及其蛋白的表达水平,较正常对照组显著升高,并随着诱导时间延长,表达水平持续升高,且屈光度变化呈负相关,与眼轴变化成正相关,这表明cochlin可能是诱发近视的始动因素或重要的早期调控因素。
[0022]
在随后的细胞实验中,重组蛋白cochlin刺激视网膜色素上皮(rpe)细胞,基因芯片和实时定量pcr分析发现rpe细胞内sfrp-1基因表达较正常对照组显著上调,同时rpe细胞释放的sfrp-1蛋白也显著增加。用cochlin刺激rpe细胞的条件培养基与猴脉络膜血管内皮细胞rf/6a孵育,可激活非经典wnt/ca
2+
/camk信号通路,但并不影响经典wnt信号通路,从而在不影响细胞增殖的条件下,促进细胞凋亡和迁移,抑制细胞的成管能力。这些结果提示,cochlin过表达可通过促进rpe细胞分泌sfrp-1,进而在脉络膜血管内皮细胞中激活非经典wnt/ca
2+
/camk信号通路,引起脉络膜血管内皮细胞功能障碍进而引起脉络膜血流灌注减少。实验同时证明,用已商业化的抑制剂way316606抑制sfrp-1,可抑制非经典wnt/ca
2+
/camk信号通路的过度激活,进而在凋亡、迁移和成管方面恢复脉络膜血管内皮细胞功能,延缓近视进展。
[0023]
继而在豚鼠的fdm模型中,经玻腔注射携带针对豚鼠cochlin基因的shrna的慢病毒颗粒,或经眼表滴加sfrp-1的抑制剂way316606,在造模后1周和6周时,这两种干预手段均可显著抑制fdm诱导的屈光度加深和眼轴延长;同时眼底光相干断层血管成像(octa)和石蜡切片形态学分析显示这两种干预手段均可以显著减轻fdm引起的脉络膜血流减少;蛋白印迹也显示cochlin shrna和way316606可以拮抗非经典wnt/ca
2+
/camk信号通路的激活。
[0024]
因此,体内、体外实验结果提示cochlin和sfpr-1在近视发病机制中起到至关重要的作用,可以作为近视对因治疗的干预靶点。
[0025]
虽然cochlin和sfrp-1两种蛋白均可作为近视干预的潜在靶点,但由于cochlin蛋白结构复杂,当前难以作为药物设计的靶点。sfrp-1是一种分泌型糖蛋白,属于分泌型卷曲相关蛋白家族的成员。在多数情况下,sfrp-1与wnt卷曲蛋白受体有部分同源结构,因此,sfrp-1可直接与wnt蛋白结合,从而竞争性抑制受体与wnt蛋白结合,进而阻止wnt信号的传递。因此,本发明中通过制备可靶定非经典wnt/ca
2+
/camk信号通路中sfrp-1蛋白的化合物来达到防治近视的效果。
[0026]
相对于现有技术,本发明所述的酰胺类化合物在制备防治近视的药物中的应用具有以下优势:
[0027]
本发明提供了如式(ⅰ)所示的化合物在制备防治近视的药物中的应用,式(ⅰ)所示化合物可以靶定非经典wnt/ca
2+
/camk信号通路中sfrp-1蛋白,是一种对sfrp-1蛋白亲和力强,且作用效果明显sfrp-1特异性抑制剂,通过与sfrp-1蛋白的活性位点进行靶向结合,能够有效拮抗sfrp-1引起的细胞内ca
2+
浓度升高,从而抑制非经典wnt/ca
2+
/camk信号通路的过度激活,进而在凋亡、迁移和成管方面恢复脉络膜血管内皮细胞功能,达到有效防治近视的效果,具备成为新的防治近视的药物的潜力,具有良好的市场化前景。
附图说明
[0028]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0029]
图1为sfrp-1蛋白一级序列(aah36503.1)模建后三维结构图;
[0030]
图2为sfrp-1蛋白结构的拉氏图(ramachandran图);
[0031]
图3中,a为同源蛋白frizzled 8的crd和xwnt8相互作用的复合物结构,b为way316606系列化合物通过盲法在sfrp-1表面的对接情况图,c为4f0a与sfrp-1三维结构叠合的远距离视图,d为4f0a与sfrp-1三维结构叠合的近距离视图;
[0032]
图4为式(ⅰ)所示化合物叠合在sfrp-1结合位点的构象图;
[0033]
图5为式(ⅰ)所示化合物在脉络膜血管内皮细胞中的活性检测结果图;
[0034]
图6为式(ⅰ)所示化合物在细胞内信号通路激活检测结果图。
具体实施方式
[0035]
除有定义外,以下实施例中所用的技术术语具有与本发明所属领域技术人员普遍理解的相同含义。以下实施例中所用的试验试剂,如无特殊说明,均为常规生化试剂;所述实验方法,如无特殊说明,均为常规方法。
[0036]
下面结合实施例及附图来详细说明本发明。
[0037]
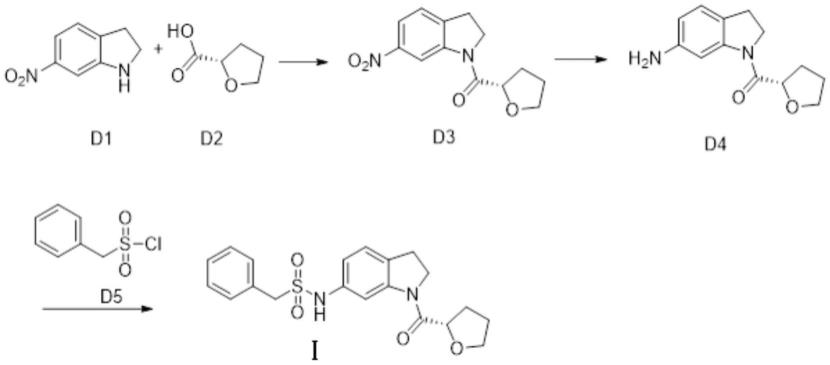
一、制备式(ⅰ)所示化合物,其合成路线如下所示:
[0038][0039]
1)中间体d3的合成
[0040][0041]
称取化合物d2(696mg,6.0mmol),加入20ml二氯甲烷,再加入草酰氯(2.16g,18.0mmol),再滴加一滴dmf,常温下继续搅拌反应2小时,体系变澄清,将体系减压浓缩干,再加入15ml二氯甲烷,使其溶解,得到d2的酰氯二氯甲烷溶液,待用;
[0042]
称取化合物d1(656mg,4.0mmol),三乙胺(1.62mg,16.0mmol),加入20ml二氯甲烷,搅拌,降温0℃以下,将上述的d2的酰氯二氯甲烷溶液滴加到体系中,滴加过程中控温0℃以下,加毕,升温至室温继续反应2小时,tlc检测,反应完毕。加入10ml水淬灭反应,分出有机相,干燥有机相,浓缩,柱层析得到化合物d3:714mg,收率68%。
[0043]
2)中间体d4的合成
[0044][0045]
称取中间体d3(600mg,2.29mmol),加入10ml甲醇,10%pd/c 100mg,用氢气置换3次,氢气氛围下常温搅拌反应过夜,次日,tlc检测反应完毕,过滤除去pd/c,浓缩,所得产品不经纯化直接下一步。
[0046]
3)化合物d的合成
[0047][0048]
将步骤2)所得的产品中间体d4加入干燥的乙腈10ml,加入三乙胺(700mg,6.87mmol),冰浴,降温到0℃以下,称取化合物d5(652mg,3.44mmol),继续冰浴下反应3小时,tlc检测,反应完毕,滴加3ml水,继续搅拌反应20分钟淬灭反应,加入50ml乙酸乙酯搅拌,分液,干燥有机相,过滤,浓缩,柱层析得到目标产物式(ⅰ)所示化合物:362mg,收率41%。
[0049]
二、效果验证
[0050]
1、sfrp-1三维结构确定
[0051]
利用蛋白结构同源模建平台swiss-model,alphafold及rosettafold的预测,结合sfrp-3的crd区域晶体结构数据。采用同源模建方法,以5xgp(pdb code)晶体结构为模板,结合100ns的分子动力学采样,sfrp-1蛋白一级序列(aah36503.1)模建后三维结构如图1所示的结构骨架图(backbone),分为ntd与crd两个结构域;图2为蛋白结构的拉氏图(ramachandran图)。
[0052]
2、sfrp-1活性位点的确定
[0053]
如图3所示,蛋白结构中所有crd区域以白色分子表面显示,xwnt8以紫色ribbon表示,sfrp-1的ntd区域以棕色ribbon表示。图3a:frizzled8结合xwnt8复合物结构信息(pdb code:4f0a)(crystal structure of xwnt8in complex with the cysteine-rich domain of frizzled 8),借鉴蛋白与蛋白相互作用(protein-protein interaction,ppi)抑制剂开发经验,我们预测与frizzled8同源的sfrp-1的crd区可能为配体的结合区域(如a中所示凹槽分);图3b:文献报道的way316606系列化合物通过盲法对接方法(blind docking)寻找位点,如图b中所示,即多个小分子占据位置,在对接中,共37个分子设定产生37*3=111个对接构象,其中有99个构象(接近90%)靶向此位点,剩余构象靶向ntd的表面,可见此位置
是sfrp-1活性位点的可能性最大;图3c,3d:是通过计算软件对sfrp-1的表面位点预测结果,其中图3c为4f0a与sfrp-1三维结构叠合的远距离视图,图3d为4f0a与sfrp-1三维结构叠合的近距离视图,图中红白小球显示区域为打分最好的潜在活性位点;4f0a结构中的frizzled 8部分与sfrp-1的crd区三维结构叠合后发现,sfrp-1的ntd区域(棕色)并没有影响crd(白色表面)区域与xwnt8蛋白的作用(紫色),表明一旦sfrp-1的活性位点被配体分子占据后,将会干扰sfrp-1与wnt蛋白的结合,非经典wnt/ca
2+
信号通路将可能受到抑制。
[0054]
3、式(ⅰ)所示化合物叠合在sfrp-1结合位点的构象图如图4所示,由图4可以看出,式(ⅰ)所示化合物不仅在结合能量上远远超过way316606,而且几何形状匹配完美。
[0055]
4、体外细胞实验
[0056]
基于已有的体外细胞实验技术,选取sfrp-1的抑制剂way316606作为阳性对照,对式(ⅰ)所示化合物的测试实验采用如下技术方案:
[0057]
1)细胞培养
[0058]
将购自中国科学院典型培养物保藏委员会细胞库(中国上海)的rf/6a细胞(猴脉络膜血管内皮细胞系)培养于含有10%fbs,100u/ml青霉素,100μg/ml链霉素和2mm l-谷氨酰胺的dmem/f12的培养基,置于37℃,5%co2条件的细胞培养箱内。
[0059]
2)细胞处理及实验分组
[0060]
将复苏后第2-3代的rf/6a细胞(4
×
104细胞/孔)种入白色非透明96孔板中,将细胞分为6组(n=3/组):
[0061]
1.正常对照组:完全培养基;
[0062]
2.sfrp-1组:完全培养基含30ng/ml sfrp-1;
[0063]
3.sfrp-1+10ⅰ组:完全培养基30ng/ml sfrp-1+10μm式(ⅰ)所示化合物;
[0064]
4.sfrp-1+2ⅰ组:完全培养基30ng/ml sfrp-1+2μm式(ⅰ)所示化合物;
[0065]
5.sfrp-1+0.4ⅰ组:完全培养基30ng/ml sfrp-1+0.4μm d式(ⅰ)所示化合物;
[0066]
6.sfrp-1+way316606组:完全培养基30ng/ml sfrp-1+2μmway316606;
[0067]
3)细胞内ca
2+
的活性测定
[0068]
孵育21小时后,吸出培养基,每孔加入100μl含10%cck-8(dojindo laboratories,kumamoto,japan)的完全培养基,37℃,5%co2条件的细胞培养箱内孵育1小时,酶标仪(tecan group ltd.,switzerland)测量450nm吸光度,代表每孔的细胞数目。然后根据fluo-8medium removal calcium assay kit的说明书,测量细胞内ca
2+
的活性。然后用每孔细胞od450nm的数值校正每孔细胞内的ca
2+
活性,最后,细胞内的ca
2+
活性用正常对照组的百分数来表示。
[0069]
式(ⅰ)所示化合物在脉络膜血管内皮细胞中的活性检测结果如图5所示。rf/6a(猴脉络膜血管内皮细胞)细胞种在96孔板中,直接加入sfrp-1(30ng/ml)重组蛋白,sfrp-1在这一浓度可以激活脉络膜血管内皮细胞内的非经典wnt/ca
2+
信号通路,同时加上三种不同浓度(10μm,2μm,0.4μm;药物剂量爬坡)的新合成的sfrp-1抑制剂化合物(ⅰ);way316606采用前期研究的最佳浓度(2μm),作为阳性对照。孵育19小时之后,用fura-8钙离子检测试剂盒检测每孔细胞内钙离子的浓度,并用每孔细胞数量(od450的读数为代表)进行校正。虚线代表正常组细胞内的钙离子水平。可见在sfrp-1的刺激下,细胞内钙离子浓度升高;而加入不同浓度的同一个药物(10μm,2μm,0.4μm),可以使细胞内ca
2+
浓度呈现出一个开口向下的
抛物线。其中,0.4μm的化合物(ⅰ)可使细胞内的钙离子浓度恢复到非常接近正常的水平。这表明新合成的化合物(ⅰ)可拮抗sfrp-1引起的细胞内ca
2+
浓度升高。
[0070]
4)蛋白印迹法确定细胞内信号通路的激活
[0071]
根据上述细胞内ca
2+
的活性测定的结果,将细胞复苏后第2-3代的rf/6a中在6孔板中(1
×
106细胞/孔),将细胞分为5组:
[0072]
1.正常对照组:完全培养基;
[0073]
2.sfrp-1组:完全培养基含30ng/ml sfrp-1;
[0074]
3.sfrp-1+0.4ⅰ组:完全培养基30ng/ml sfrp-1+0.4μm d式(ⅰ)所示化合物;
[0075]
4.sfrp-1+dmso组:完全培养基30ng/ml sfrp-1+dmso(1/5000体积比);
[0076]
5.sfrp-1+way316606组:完全培养基30ng/ml sfrp-1+2μm
[0077]
way316606;
[0078]
依照组别对细胞进行刺激21小时后,根据蛋白酶抑制剂与中性ripa裂解液=1∶99的比例,配制组织蛋白裂解液,6孔板每孔加300μl蛋白裂解液,充分裂解细胞。取出细胞裂解液,于冰上孵育20min,12,000rpm,低温离心20min,收集上清液于新的1.5ml离心管内。
[0079]
根据bca蛋白定量试剂盒(康为世纪,北京)操作说明,首先,稀释bsa标准品,接着,用ripa中性裂解液5倍稀释蛋白样本,并将标准品和稀释后蛋白样本各25μl分别加入96孔板内。然后,将bca试剂盒中的a液和b液按50:1的体积比,配制bca工作液,充分混匀。每孔加入200μlbca工作液,37℃孵育30min,待其冷却至室温后,用酶标仪(tecan group ltd.,switzerland)混匀震荡30s,并在562nm处测定吸光值,最后绘制标准曲线,计算各样品中蛋白浓度值。
[0080]
将蛋白样品(50μg/孔)100℃,10min高温变性后,加入适量4x蛋白上样缓冲液混合,加入梳孔内,进行sds-page电泳,恒压100v,当指示剂到达胶板底部时停止电泳。将胶从玻璃板内取下,去掉上层浓缩胶,将相应大小的pvdf膜在甲醇中浸泡15s后,取出并浸泡于去离子水中2min,然后从负极到正极按照滤纸-胶-膜-滤纸的顺序,放入转膜支撑夹内,最后将夹子放入转膜槽内,倒入转膜液,在低温的条件下进行恒压100v转膜100min。
[0081]
转膜完毕后,将pvdf膜在1x tbst缓冲液内漂洗3次,每次快速摇床10min,最后用5%的脱脂奶粉室温封闭2h。封闭结束后,将膜分别与封闭液配制的抗camkⅱ(兔单抗:1:1000,cell signaling technology,美国),抗phospho-camkⅱ(兔多抗:1:1000,cell signaling technology,美国)和抗β-actin(β-actin鼠单抗:1:5000,abcam,美国)的一抗,在4℃摇床缓慢孵育过夜。
[0082]
转天,用tbst洗膜3次,每次10min。随后将膜与tbst稀释的辣根过氧化物酶标记的羊抗兔(1:10000,abcam,美国)或羊抗鼠(1:5000,abcam,美国)二抗,在室温慢速摇床孵育2h。然后,tbst快速洗膜4次,每次10min,将膜取出,平铺于发光成像仪(uvp,llc,upland,ca,usa)指定位置,均匀滴加显影液(amersham biosciences,piscataway,nj,usa)以覆盖要显影的膜,利用发光成像仪(uvp,llc,upland,ca,usa)拍摄图片,使用image j软件(national institute of health,bethesda,md,usa)对条带灰度值进行定量分析。
[0083]
结果表明,sfrp-1可以使p-camkii-α和p-camkii-β的表达增高,而0.4μm的化合物(i)可使p-camkii-α和p-camkii-β的表达降至正常水平,其效果与2μm way316606相似;而溶剂对照dmso没有效果(图6)。总的camkii-α和camkii-β的表达趋势与其相应的磷酸化蛋
白相似。此实验中,p-camkii-α和总的camkii-α的变化趋势比相应的β亚型蛋白更加明显(图6)。
[0084]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1