反义核酸糖基缀合物及其制备方法和在肝癌治疗中的应用与流程
1.本发明涉及一系列反义核酸糖基缀合物及其制备方法,还涉及由所述的反义核酸糖基缀合物制成的药物制剂及其在治疗肝癌中的应用。本发明属于生物医药技术领域。
背景技术:
2.糖类衍生物通过凝集素和糖类结合蛋白参与许多生物过程,如信号传递、细胞表面识别等[chembiochem,2004,5(6):740-764.]。糖类衍生物-寡核苷酸缀合物的合成已经被深入的探讨。半乳糖已成为研究最多的糖类衍生物,用于增强寡核苷酸的靶向递送[j am chem soc,2012,134(4):1978-1981.mol ther,2017,25(1):71-78.]。n-乙酰氨基半乳糖是一个具有高亲和性的靶向基团,它能特异性结合肝脏中脱唾液酸糖蛋白受体[mol ther nucleic acids,2017,9:34-47.](asgpr),常被用于核酸药物的肝靶向递送之中[bioconjug chem,2015,26(8):1451-1455.],效果显著。尽管还有多种其他结构变体,一个典型的三簇galnac结构是核心缀合部分[nucleic acid ther,2018,28(3):109-118.]。galnac缀合可使on的效力在小鼠中提高~7倍,在人类患者中提高~30倍[nucleic acids res,2014,42(13):8796-8807.]。相对于纳米颗粒复合物,galnac缀合物尺寸较小,化学成分明确单一,合成成本低。
[0003]
galnac偶联策略在一些制药公司的药物开发中占了很大比重,如dicema pharmaceuticals、silence therapeutics、arbutus biopharma和arrowhead pharmaceuticals均在研发galnac缀合相关产品[trends pharmacol sci,2021,42(7):588-604.]。其中最突出的是alnylam,目前上市的三款galnac缀合药物均来自该公司,分别用于治疗急性肝卟啉症、高胆固醇血症和高草酸尿症[nat rev drug discov,2020,19(10):673-694.]。首个galnac缀合sirna givosiran于2019年获得fda批准用于治疗成人ahp(acute hepatic porphyria,急性肝卟啉症),ahp是一种罕见的遗传性疾病,是由于血红素生成过程中代表性酶的缺陷致使患者体内卟啉代谢产物蓄积引起神经性内脏症状[n engl jmed,2019,380(6):549-558.]。givosiran的原理为诱导编码aminolevulinate synthase 1的基因沉默,减少ala和胆色素原的生成,缓解卟啉或其前体的蓄积。givosiran为21/23个核苷酸长度,其采用了ps,2
′‑
f和2
′‑
ome的化学修饰,iii期临床结果显示其可以显著降低ahp的发生次数,并减少氯化血红素(治疗贫血)的使用[expert opin biol ther,2013,13(3):429-435.]。此外,其他制药巨头公司也有自己的galnac缀合on生产管线,并处于不同的临床研究阶段。它们具有的共同特点是,都以至少三簇连续的糖分子结构出现。若只有单个galnac基团,文献报道活性会下降一倍以上,靶标亲和力会下降10倍左右[.j med chem,2016,59(6):2718-2733.]。adrien[mol ther nucleic acids,2020,21:242-250.]等人系统研究了galnac缀合分子个数,缀合位点的活性差异发现,至少需要连续两个galnac分子才能实现一定的活性水平,三个最为合适,再增加galnac分子个数则不会有明显的活性提升。此外,在sirna缀合位点的选取上,除了反义链的5
′
端缀合会明显影响活性外,其他三个位点的缀合均可取得不错的疗效。另外一点值得注意的是,在正义链的两端分别缀合
单个galnac分子,在体内观察到了更显著的基因沉默,这也意味着大大简化了galnac缀合前体的合成难度。
[0004]
显而易见,这种以受体介导的递送方式适用于绝大多数肝部疾病的治疗。galnac缀合物的临床研发管线涉及多种疾病模型,如代谢性疾病、心血管疾病、传染性疾病及遗传性疾病等[acs omega,2021,6(25):16259-16265.methods mol biol,2021,2282:57-75.]。brii-835(vir-2218)是由alnylam/vir/腾盛博药合作开发的一种经皮下注射给药的靶向hbv的galnac-sirna药物,其介导rna干扰,具有对hbv直接抗病毒活性和诱导有效免疫应答的潜力。它是首个进入临床的采用增强型稳定化学+(esc+)技术的sirna,可增强稳定性并最大程度地降低脱靶效应,从而可提升治疗效力。小鼠实验单次皮下给药3mg/kg,沉默10种hbv基因型的cccdna和整合dna的所有hbv转录物。临床2期皮下第1天和第29天2次给药(20~200mg/次),24周hbeag阴性和hbeag阳性患者及在所有剂量下均观察到hbsag显著剂量依赖和持久降低,慢性乙肝患者耐受良好(easl2021)。然而,针对于肝癌的治疗,galnac缀合似乎不是一个良好的选择。因asgpr受体在各种分化的肝肿瘤细胞中都不同程度的低表达,可能会严重影响药物的摄取。但是也有观点认为asgpr受体在肝细胞表面及其丰富,即使部分低表达,在正常给药浓度下,剩余的受体量足够介导药物入胞。该配体与小分子药物的偶联在治疗肝细胞癌方面也取得了不错的进展[mol pharm,2021,18(1):461-468.]。ionis公司试图将galnac偶联技术推广至肝癌的治疗上,在体内外的靶基因沉默上均可实现较未缀合分子相对较好的效果[mol ther,2019,27(9):1547-1557.],但是在该公司后续的一项类似实验中,在体内galnac偶联并未显示出更优的基因沉默活性,其原因还是归结于asgpr在高度分化的人hcc肿瘤组织中表达下调[cancerres,2022,82(5):900-915.]。
[0005]
此外,其他单糖或者多糖分子利用细胞膜上的受体作为靶向用于药物传递的研究屡见不鲜。肿瘤表现出增强的葡萄糖摄取特性,以维持其快速的细胞增殖。利用此特性,patra等人[angew chem int ed,2016,55(7):2550-2554.]设计合成了一系列葡萄糖-顺铂偶联物来增强肿瘤细胞的摄取,同时降低顺铂药物带来的毒副作用。体外研究证明,与正常上皮细胞相比,葡萄糖-顺铂偶联物可通过葡萄糖转运蛋白,优先在肿瘤细胞中积聚并杀死肿瘤细胞。甘露糖受体是先天免疫系统中一个重要的模式识别受体,主要存在于巨噬细胞和树突状细胞的细胞膜表面,在维持内稳态、识别病原和抗感染等过程中都发挥着重要作用。以此受体为靶点开发甘露糖相关靶向制剂也是研究的一个热点[adv funct mater,2018,28(30):1802227.bioact mater,2020,6(3):697-711.]。此外,也有研究报道氨基葡萄糖,甘露糖等分子自身可直接或间接的诱导肿瘤细胞凋亡[chem biol interact,2019,300:63-72.cancer,2018,124(16):3307-3318.nature,2018,563(7733):719-723.]。
[0006]
王升启等人研发的抗肝癌反义核酸ct102已完成全部的临床前研究,于2021年2月进入临床研究i期,其以人胰岛素样生长因子i型受体(igf1r)的mrna为靶标,疗效明确且安全性高,但给药浓度及给药频次较高(小鼠水平给药~10mg/kg,隔日给药,累计给药10次尾静脉注射)。研究发现igf1r及其配体在急性白血病、多发性骨髓瘤、乳腺癌、前列腺癌、卵巢癌、子宫内膜癌、宫颈癌、非小细胞肺癌、尤因肉瘤等恶性肿瘤中均表达异常。进一步研究证明igf1r表达与肿瘤的生长、转移、放化疗治疗抵抗密切相关。igf1r经由多种信号转导通路介导了肿瘤细胞的恶性化增殖、侵袭以及转移过程,同时还介导了肿瘤血管的生成以及肿瘤细胞抗凋亡等作用。igf1r主要由igf1激活,它也可以与igf2和胰岛素结合,结合胰岛素
样生长因子(igf1或igf2)配体后,通过自身磷酸化,激活p磷脂酰肌醇3激酶(pi3k),akt,rac和促分裂原活化蛋白激酶(mapk)途径。这些途径调节参与各种细胞功能的关键基因,例如增殖,存活,分化,转化和凋亡[world j hepatol,2014,6(10):716-737cancer,2012,118(16):3993-4003]。多年来的研究已经证明igf1r在肿瘤发生和转移中发挥不可替代的作用,是目前公认的肿瘤靶基因之一[expert opin inv drug,2019,28(12):1101-1112clin cancer res,2015,21(19):4270-4277]。在乳腺癌患者中,igf1r通路与雌激素受体(er)和表皮生长因子受体2(erbb2)信号通路密切相关,igf1r位于pi3k-akt1-mtor通路的上游,超过一半的乳腺癌患者pi3k-akt1-mtor通路被异常激活[cancerres,2011,71(21):6773-6784];肉瘤-肿瘤模型的临床前数据表明,igf1r通路在尤因肉瘤和横纹肌肉瘤患者的肿瘤生长、转移和血管生成中特别重要,igf1r抑制剂已在这些肿瘤患者中初步应用[lancet oncol,2010,11(2):129-135];igf1r蛋白在非小细胞肺癌(nsclc)细胞系和患者样本中的表达水平在腺癌和鳞状组织中都很高,igf1r表达与nsclc患者的不良预后相关[thoraccancer,2020,11(4):875-887]。尽管如此,igf1r在不同肿瘤中表达紊乱的调控机制依然不甚明确,迄今为止还未有针对该靶点的核酸药物上市。
[0007]
本发明发明人前期研究结果证明了dnca/cld/peg/ct102药物制剂在体内的安全性与有效性(3.75mpk/2天一次/iv),本研究将反义核酸与具有靶向作用的糖配体结合,构建出一系列反义核酸ct102
moe5
缀合物,并同样通过dnca/cld/peg递送体系包载转染,实现了低剂量下(2mpk/4天一次/iv)优越的抗肿瘤增殖效果。
技术实现要素:
[0008]
本发明的目的之一是提供一种反义核酸糖基缀合物及其制备方法;
[0009]
本发明的目的之二是提供一种反义核酸联合包载加末端缀合的组合递送策略;
[0010]
本发明的目的之三是提供由所述的含有反义核酸药物的脂质复合物制备得到的药物制剂及其在治疗原发性肝癌中的用途。
[0011]
为了达到上述目的,本发明采用的技术方案如下:
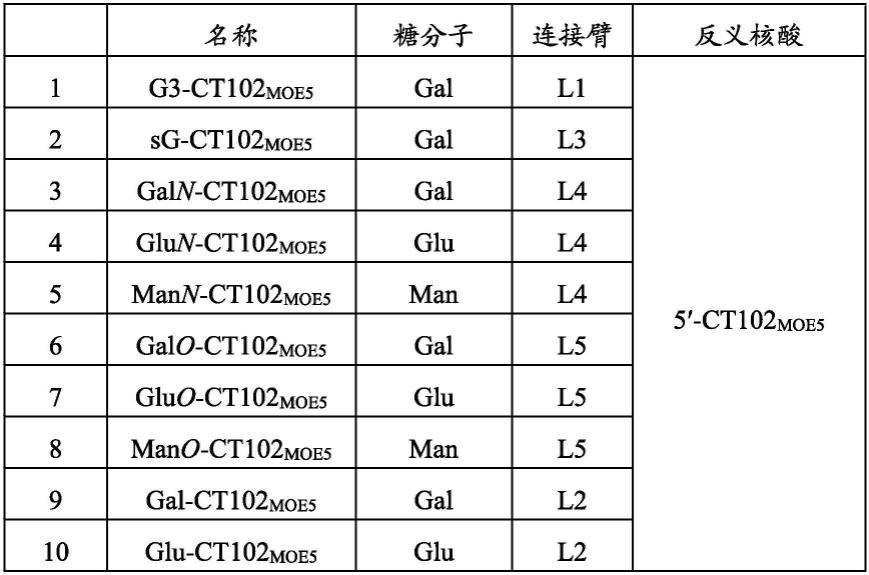
[0012]
本发明一种反义核酸糖基缀合物,所述的反义核酸糖基缀合物是由糖分子通过连接臂缀合于反义核酸5
′
末端共价偶联而成;
[0013]
其中,所述的糖分子为乙酰氨基半乳糖(gal),乙酰氨基葡萄(glu)或甘露糖(man);
[0014][0015]
其中,所述的连接臂的结构如下l1、l2、l3、l4或l5所示;
[0016][0017]
其中,所述连接臂左端与糖分子连接,右端与反义核酸5
′
端羟基连接。
[0018]
其中,优选的,当所述的糖分子为乙酰氨基半乳糖时,连接臂的结构如l1,l2,l3,l4或l5所示,当所述的糖分子为乙酰氨基葡萄时,连接臂的结构如l2,l4或l5所示,当所述的糖分子为甘露糖时,连接臂的结构如l2,l4或l5所示。
[0019]
其中,优选的,其中的反义核酸选自以下序列:
[0020]
ct102:tscscs tscscs gsgsas gscscs asgsas cststs csa
[0021]
ct102
moe5
:tesmces
mm
cestesmcesmcsgsgsasgsmcsmcsasgsasmcestestesmcesae;
[0022]
phn01:gsasgs cscsasgsascststscs aststscscststst
[0023]
phn02:gsasgs asasascsasgs gsasgscscscscscsascsa
[0024]
phn02
moe5
:gesaesgesaesaesasmcsasgsgsasgsmcsmcsmcsmcesmcesaesmcesae
[0025]
phn03:cscsgsgsasgscscsasgsascststscs aststscsc
[0026]
phn04:tstscstscscsascstscsgstscsgsgscscsasgsa
[0027]
phn05:gststscscscsgscsasgstsgs gscscsgscsgsgsg
[0028]
phn06:gscstsgscstsgsastsasgstscsgststsgscsgsg
[0029]
phn07:tscscscstsgsgsascsascsasgsgstscscscscsa
[0030]
其中:mc表示5-甲基胞嘧啶修饰(5mc);s表示磷硫代修饰(ps);e表示2
′‑
o-moe修饰(2
′‑
o-moe);结构分别如下所示:
[0031][0032]
其中,优选的,所述的反义核酸糖缀合物选自以下化合物:
[0033][0034]
其中,优选的,还包括在所述的反义核酸糖基缀合物的反义核酸的3
′
端进一步缀合荧光标记分子。
[0035]
其中,优选的,缀合荧光标记分子的反义核酸糖基缀合物的结构如下所示:
[0036][0037]
进一步的,本发明还提出了所述的反义核酸糖基缀合物在制备治疗肿瘤的药物制剂中的应用,优选的,所述的肿瘤包括肝癌。
[0038]
更进一步的,本发明还提出了一种用于抗肝癌的药物制剂,所述的药物制剂中包括本发明所述的反义核酸糖基缀合物与dnca、cld以及dspe-peg形成的脂质复合物,其中所述的dnca、cld、dspe-peg的结构如下:
[0039][0040]
其中,优选的,所述的dnca、cld、dspe-peg与反义核酸糖基缀合物中反义核酸物质的量之比为30∶30∶0.6∶1、20∶20∶0.4∶1、40∶20∶0.6∶1或20∶40∶0.6∶1。
[0041]
再进一步的,本发明还提出了所述的药物制剂在制备治疗原发性肝癌药物中的用途。
[0042]
相较于现有技术,本发明的优点在于:
[0043]
1.本发明通过在前期实验证明的抗肝癌活性最优的抗肝癌反义核酸序列ct102
moe5
的基础上构建了一系列其5
′
末端糖基缀合物,体内外实验证明其具有优异的血清稳定性及体内半衰期,单次静脉给药后药物可在体内蓄积40天左右。同时,糖分子通过细胞表面的糖基转运体增加了药物的摄取能力。通过不同连接臂及糖分子的筛选,确定了部分活性较好的候选物进行了体内药效实验。
[0044]
2.各反义核酸缀合物在dnca/cld/dspe-peg包载下可形成稳定的150nm左右的球形纳米颗粒。此制剂在2mpk/4天一次的静脉给药方案下,显示出了良好的体内肿瘤抑制效果,相较于原始未修饰链ct102,进一步降低了给药频率及给药剂量,安全性良好。其中glu-ct102
moe5
在实验中表现出稳定的最优的抗肝癌活性,可作为进一步研究的抗hcc反义核酸候选物。
[0045]
3.本发明提供了一系列针对于ct102靶标igf1r mrna的反义核酸新序列,其分别靶向igf1r mrna外显子的不同区域。通过细胞水平的活性筛选,优选出phn02与phn07,其对肿瘤细胞增殖抑制、靶基因沉默活性以及促凋亡租用与原始序列ct102相比更具优势。gapmer化学修饰得到的phn02
moe5
体内外活性进一步提升,明显优于非修饰物phn02及ct102。体内抗肿瘤活性实验表明phn02
moe5
与实验室前期优选的ct102
moe5
具有相当的抑制肿瘤生长效果,值得进一步临床研发。
附图说明
[0046]
图1为g3ac-nhs的合成路线;
[0047]
图2为sgalnac(sg)亚磷酰胺单体的合成路线;
[0048]
图3为gal(n/o),glu(n/o),man(n/o)缀合前体的合成路线;
[0049]
图4为gal/glu/man缀合前体活泼酯的合成路线;
[0050]
图5为液相溶液汇聚法合成g3-on流程图;
[0051]
图6为溶液汇聚法合成cy5.5-g3-on荧光标记物流程图;
[0052]
图7为丙烯酰胺凝胶电泳考察ct102修饰及缀合物在50%fbs中的稳定性;
[0053]
其中每孔核酸样品为10pmol;
[0054]
图8为流式细胞术检测脂质体包载ct102各修饰、缀合物复合物在hepg2、huh7中4h摄取情况;
[0055]
其中各核酸为cy5.5标记,其浓度为200nm,dnca/cld/aso=30/30/1;紫色吸收峰代表基底,棕色吸收峰代表样品;
[0056]
图9中a为tem电镜观察dnca/cld体系包载ct102
moe5
各修饰物/缀合物的亚显微结构(100nm尺度);b为dnca/cld体系包载ct102
moe5
各修饰物/缀合物电位及粒径(n=3);
[0057]
图10为dnca/cld/dspe-peg包载的cy5.5标记ct102
moe5
和g3-ct102
moe5
体内分布及荧光定量分析结果(静脉给药);
[0058]
其中,a.ct102
moe5
或g3-ct102
moe5
标记物在包载及裸给情况下,不同时间点(2h,12h,24h,2d,3d,6d,9d,15d,21d,28d,35d,40d)活体成像结果;b.各组小鼠全身荧光定量随时间变化曲线。数据使用mean
±
sd表示,n=3;
[0059]
图11为dnca/cld/dspe-peg包载的cy5.5标记g3-ct102
moe5
及ct102
moe5
离体组织荧光定量分析结果;
[0060]
其中,a.不同时间点(4h,1d,2d,5d,10d,15d)各组小鼠心,肺,肝,脾,肾,肠荧光成像图;b.不同时间点(4h,1d,2d,5d,10d,15d)各组小鼠心,肺,肝,脾,肾,肠荧光强度占比情况(n=3);
[0061]
图12为各缀合物对hepg2、huh7细胞增殖抑制活性及靶标igf1r mrna沉默活性(100nm)。*代表p<0.05;
[0062]
图13为dnca/cld/peg包载的ct102修饰物及缀合物体内抗肿瘤效果。
[0063]
其中,a.各组小鼠肿瘤增殖倍数概况;b.给药后各组小鼠体重情况;d.给药后28天相较于给药前肿瘤增长倍数情况;c.给药后不同时间点各组小鼠肿瘤生长曲线。*代表p<0.05,***代表p<0.001;
[0064]
图14为靶向igf1r的反义核酸新序列在hepg2和huh-7细胞水平药效;
[0065]
其中,a.mix包载反义核酸序列对hepg2和huh-7细胞增值抑制活性;b.mix包载反义核酸序列对hepg2和huh-7细胞中igf1r mrna沉默效果。*代表p<0.05;
[0066]
图15为流式细胞术考察混合脂材mix包载ct102、phn02、phn07在hepg2中促凋亡能力;
[0067]
其中,blank:空白溶剂对照;nc:mix包载乱序aso;ct102、phn02、phn07分别为mix包载反义核酸ct102、phn02、phn07制剂。
[0068]
图16为靶向igflr的反义核酸新序列对a549、mcf-7、b-cpap细胞的增殖活性及靶基因沉默效果;
[0069]
其中,a.mix包载反义核酸序列对a549、mcf-7和b-cpap细胞增值抑制活性;b.mix包载反义核酸序列对a549、mcf-7和b-cpap细胞中igf1r mrna沉默效果。
[0070]
图17为phn02
moe5
制剂在hepg2和huh-7细胞水平药效;
[0071]
其中,a.mix包载反义核酸phn02的2
′‑
o-moe修饰物phn02
moe5
对hepg2和huh-7细胞增值抑制活性;b.mix包载反义核酸phn02的2
′‑
o-moe修饰物phn02
moe5
对hepg2和huh-7细胞中igf1r mrna沉默效果。
[0072]
图18为mix包载反义核酸phn02
moe5
对原位组织移植瘤肝癌小鼠的药效情况;
[0073]
其中,a.抗肝癌反义核酸治疗原位组织移植瘤肝癌小鼠的给药方案;b.各组给药
后不同时间点肿瘤荧光强度与给药前肿瘤荧光强度比值增长情况;c.给药后28天肿瘤荧光强度与给药前肿瘤荧光强度比;d.各组小鼠肿瘤组织的igf1r mrna表达情况(n=4);e.小鼠给药期间体重变化情况;**代表p<0.01,****代表p<0.0001;
[0074]
图19为mix包载反义核酸phn02
moe5
对原位组织移植瘤肝癌小鼠的药效实验不同阶段肿瘤成像图。
具体实施方式
[0075]
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随具体实施例的描述而更为清楚。但实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。
[0076]
第一部分缀合物前体结构的构建合成
[0077]
实施例1 g3ac-nhs的合成
[0078]
g3ac-nhs的合成路线如图1所示。
[0079]
称取galnac羧酸原料300mg(0.15mmol),n-羟基丁二酰亚胺(nhs)(25.8mg,0.23mmol),1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci)(43.2mg,0.23mmol)溶解于5ml无水二氯甲烷(dcm)中,室温(rt)下反应12h。饱和nahco3水溶液洗涤两次,饱和食盐水洗涤一次,无水na2so4干燥。后旋蒸干燥,得g3ac-nhs白色固体产物270mg,产率85.3%。1h nmr(400mhz,dmso)δ7.87-7.70(m,9h),5.75(s,1h),5.21(d,j=3.4hz,3h),4.97(dd,j=11.2,3.4hz,3h),4.49(d,j=8.4hz,3h),4.06-3.98(m,9h),3.87(dt,j=11.2,8.8hz,3h),3.71(dt,j=9.5,5.6hz,3h),3.59-3.49(m,12h),3.41(dt,j=9.5,6.0hz,3h),3.03(p,j=6.4,6.0hz,12h),2.81(s,4h),2.65(t,j=7.2hz,2h),2.28(t,j=6.3hz,6h),2.10(s,9h),2.04(t,j=7.1hz,8h),2.00(s,9h),1.89(s,9h),1.77(s,9h),1.60(q,j=7.2hz,2h),1.53-1.40(m,20h),1.37-1.31(m,2h),1.28-1.21(m,10h).esi-msfor c
95h151n11o41
[(m+2h)/2]
+
found 1052.6102 calcd 1052.5051.
[0080]
实施例2其他单糖缀合物的合成
[0081]
1.2.1 sgalnac(sg)亚磷酰胺单体的合成
[0082]
sgalnac(sg)亚磷酰胺单体的合成路线如图2所示。
[0083]
化合物2的合成
[0084]
取d-(+)-半乳糖胺盐酸盐(10.0g,46.37mmol),4-二甲基氨基吡啶(4-dmap)(548mg,4.6mmol),溶于100ml无水dcm中,加入三乙胺(et3n)(38ml,278mmol),冰浴条件下,向反应体系中缓慢滴加乙酸酐(ac2o)(36ml,340mmol)。温度升至室温,搅拌反应24小时。抽滤得到固体,再次用乙酸乙酯浸泡洗涤抽滤两次,得纯产物。干燥,称重,得到白色粉末状化合物2(15.36g,39.47mmol),产率85.1%。1h nmr(400mhz,dmso)δ7.89(d,j=9.2hz,1h),5.65(d,j=8.8hz,1h),5.27(d,j=3.4hz,1h),5.07(dd,j=11.3,3.4hz,1h),4.27-4.18(m,1h),4.15-3.96(m,3h),2.12(s,3h),2.04(s,3h),1.99(s,3h),1.91(s,3h),1.78(s,3h).esi-ms for c
16h23
no
10
[m+h]
+
found 390.1449,calcd 390.1395;[m+na]
+
found412.1272,calcd 412.1215.
[0085]
化合物3的合成
[0086]
称取化合物2(10g,25.7mmol)溶于无水100ml dcm中,氩气保护,冰浴条件下缓慢滴加三氟甲磺酸三甲基硅酯(10ml,51.7mmol),30℃反应3小时,冰浴下滴加三乙胺(14.4ml,103.4mmol),搅拌20min。蒸干溶剂柱层析分离,ea∶pe=2∶1。分离得透明油状物化合物35.76g,产率68.1%,如图2。1h nmr(400mhz,cdcl3)δ5.87(d,j=6.8hz,1h),5.32(t,j=3.1hz,1h),4.78(dd,j=7.3,3.4hz,1h),4.15-4.10(m,1h),4.09-3.96(m,2h),3.87(td,j=7.0,1.4hz,1h),1.99(s,3h),1.95-1.90(m,9h).esi-ms for c
14h19
no8[m+h]
+
found 330.1257,calcd330.1184.
[0087]
化合物5的合成
[0088]
化合物3(1.00g,3.04mmol),6-苄氧羰酰氨基-1-己醇(化合物4)(1.53g,6.08mmol)溶于10ml二氯乙烷中,滴加三甲硅基三氟甲磺酸脂(tmsotf)(0.56ml,3.04mmol),氩气保护,35℃反应6小时。加入dcm稀释反应液,倒入冰的饱和nahco3水溶液淬灭反应,洗涤两次,饱和食盐水洗涤一次,合并有机相,无水na2so4干燥。蒸干溶剂,柱层析分离,ea∶pe=2∶1。得白色固体化合物50.87g,产率49.3%。1h nmr(400mhz,dmso)δ7.81(d,j=9.2hz,1h),7.39-7.30(m,5h),7.22(t,j=5.8hz,1h),5.21(d,j=3.4hz,1h),5.00(s,2h),4.97(dd,j=11.2,3.4hz,1h),4.49(d,j=8.5hz,1h),4.05-3.99(m,3h),3.91-3.83(m,1h),3.73-3.66(m,1h),3.44-3.38(m,1h),2.98(q,j=6.6hz,2h),2.10(s,3h),2.00(s,3h),1.90(s,3h),1.77(s,3h),1.48-1.34(m,4h),1.28-1.21(m,4h).esi-ms for c
28h40
n2o
11
[m+h]
+
found 581.2717calcd 581.2710;[m+na]
+
found 603.2537,calcd603.2530.
[0089]
化合物7的合成
[0090]
称取己二酸5g(34.21mmol),edci 19.67g(102.63mmol),nhs(11.8g,102.63mmol)于250ml茄形瓶中,加入无水dcm溶解,室温反应6小时,析出白色固体。滤除反应液,滤饼用dcm洗涤两次,得白色粉末状化合物77.49g(22.01mmok),产率64.6%。1h nmr(400mhz,dmso)δ2.82(s,8h),2.78-2.71(m,4h),1.78-1.68(m,4h).esi-ms for c
14h16
n2o8[m+h]
+
found 341.1025calcd 341.0985;[m+na]
+
found 363.0846,calcd 363.0804.
[0091]
化合物9-2的合成
[0092]
(3r,5s)-5-羟基甲基-3-吡咯烷醇盐酸盐1.00g(6.51mmol)用乙腈溶解,加入三乙胺2.25ml(16.27mmol),冰浴下滴加三氟乙酸乙酯1.87ml(15.62mmol),反应过夜。过滤除去不溶物,滤饼用ea洗涤,合并有机相旋蒸干燥,得白色粉末状化合物9-2(粗品)1.25g(5.87mmol),产率90.1%。esi-ms for c7h
10
f3no3[m+h]
+
found 214.0701calcd 214.0691.
[0093]
化合物9-3的合成
[0094]
化合物9-21.25g(5.87mmol)溶于无水吡啶中,氩气保护下缓慢滴加吡啶溶解的4,4
′‑
双甲氧基三苯甲基氯(dmtrcl)(1.98g,5.87mmol),反应过夜,旋蒸干燥,柱层析分离,pe∶ea=6∶1,收集产物后旋蒸干燥,得黄色固体化合物9-32.71g(5.25mmol),产率89.6%。1h nmr(400mhz,dmso)δ7.34-7.16(m,9h),6.92-6.85(m,4h),5.17(s,1h),4.46(s,1h),4.35-4.25(m,1h),3.74(s,6h),3.71-3.58(m,2h),3.39(dd,j=9.5,4.0hz,1h),3.00(dd,j=9.5,2.9hz,1h),2.20-2.11(m,1h),2.00-1.89(m,1h).esi-ms for c
28h28
f3no5[m+na]
+
found 538.1835,calcd538.1817.
[0095]
化合物9的合成
[0096]
化合物9-3(2.71g,5.25mmol)用甲醇溶解稀释,搅拌下滴加koh溶液(2eq),加毕搅拌3小时,后旋蒸干燥除去部分溶剂,加水静置,过滤,滤饼用水洗涤,后用dcm复溶,无水naso4干燥,旋蒸干燥后,柱层析分离,dcm∶meoh=30∶1,得黄色固体化合物91.56g(3.72mmol),产率70.7%。1h nmr(400mhz,dmso)δ7.44-7.22(m,9h),6.95-6.86(m,4h),5.20(s,1h),4.31(s,1h),3.75(s,6h),3.73-3.72(m,1h),3.24-3.07(m,3h),2.95(d,j=11.9hz,1h),1.85(dd,j=13.2,6.5hz,1h),1.63-1.53(m,1h).esi-ms for c
26h29
no4[m+h]
+
found 420.2202calcd 420.2175;[m+na]
+
found 442.2021,calcd 442.1994.
[0097]
化合物8的合成
[0098]
化合物5(3g,5.17mmol),醋酸(1.5ml,25.85mmol)和钯碳(0.1eq)溶解于thf中,h2气氛下反应4小时。硅藻土过滤,旋蒸干燥得化合物6,10ml二氯甲烷复溶备用。化合物7(2.64g,7.75mmol),三乙胺(4.29ml,31mmol)溶解于二氯甲烷中,向体系中缓慢滴加溶解的化合物6,35℃反应4小时。旋蒸干燥,柱层析分离,acn∶dcm=6∶1,得透明油状化合物81.24g,产率38.7%。1h nmr(400mhz,dmso)δ7.81(d,j=9.2hz,1h),7.75(t,j=5.6hz,1h),5.21(d,j=3.4hz,1h),4.97(dd,j=11.2,3.4hz,1h),4.49(d,j=8.5hz,1h),4.09-3.97(m,3h),3.92-3.82(m,1h),3.75-3.66(m,1h),3.44-3.37(m,1h),3.01(q,j=6.6hz,2h),2.81(s,4h),2.70-2.64(m,2h),2.11(s,3h),2.10-2.05(m,2h),2.00(s,3h),1.90(s,3h),1.77(s,3h),1.62-1.54(m,j=3.5hz,4h),1.49-1.41(m,2h),1.40-1.33(m,2h),1.28-1.21(m,4h).esi-msfor c
30h45
n3o
14
[m+h]
+
found 672.3044 calcd672.2980;[m+na]
+
found 694.2867,calcd 694.2799.
[0099]
化合物10的合成
[0100]
化合物8(500mg,0.75mmol),化合物9(470mg,1.12mmol)溶解于10ml dcm中,滴加tea(0.16ml,1.12mmol),室温反应8小时。蒸干溶剂,柱层析分离,dcm∶meoh=30∶1。得黄色固体化合物10549mg,产率75.1%。1h nmr(400mhz,dmso)δ7.81(d,j=9.2hz,1h),7.75-7.69(m,1h),7.34-7.27(m,4h),7.23-7.16(m,5h),6.90-6.85(m,4h),5.21(d,j=3.4hz,1h),4.98(d,j=3.9hz,1h),4.92(dd,j=25.7,3.9hz,1h),4.48(d,j=8.4hz,1h),4.44-4.35(m,1h),4.17-4.11(m,1h),4.06-3.99(m,3h),3.91-3.83(m,1h),3.74(s,6h),3.71-3.65(m,1h),3.63-3.56(m,1h),3.42-3.37(m,1h),3.20-3.13(m,1h),3.03-2.97(m,3h),2.22(t,j=7.2hz,2h),2.10(s,3h),2.05(t,j=7.2hz,2h),1.99(s,3h),1.89(s,3h),1.77(s,3h),1.50-1.33(m,8h),1.28-1.19(m,6h).esi-ms for c
52h69
n3o
15
[m+h]
+
found 976.4871calcd 976.4807;[m+na]
+
found 998.4701,calcd998.4626.
[0101]
sgalnac亚磷酰胺(sg)的合成
[0102]
化合物10(120mg,0.12mmol),双(二异丙基氨基)(2-氰基乙氧基)膦(72.4mg,0.24mmol),1h-四氮唑(16.8mg,0.24mmol),溶解于10ml无水乙腈中,氩气保护下,室温反应4小时。旋蒸干燥,dcm复溶,使用饱和nahco3洗涤一次,饱和食盐水洗涤一次,na2so4干燥。再次旋蒸干燥,柱层析分离,dcm∶acn=20∶1。得透明油状sgalnac phosphoroamidite 98.9mg,产率70.9%。1h nmr(400mhz,dmso)δ7.81(d,j=9.3hz,1h),7.76-7.68(m,1h),7.33-7.27(m,4h),7.23-7.16(m,5h),6.90-6.84(m,4h),5.22(d,j=3.4hz,1h),4.97(dd,j=11.2,3.4hz,1h),4.49(d,j=8.5hz,1h),4.21-4.11(m,1h),4.07-3.98(m,3h),3.87(dt,j=11.3,8.8hz,1h),3.74(s,6h),3.73-3.63(m,3h),3.62-3.35(m,5h),3.04-2.93(m,3h),
2.76(q,j=5.7hz,2h),2.31-2.16(m,2h),2.10(s,3h),2.08-2.02(m,2h),1.99(s,3h),1.89(s,3h),1.76(s,3h),1.55-1.33(m,8h),1.32-1.17(m,6h),1.18-1.01(m,12h).
31
p nmr(162mhz,dmso)δ147.01-146.05(m).esi-msfor c
61h86
n5o
15
p[m+k]
+
found 1198.5721 calcd 1198.5495.
[0103]
1.2.2 gal(n/o),glu(n/o),man(n/o)缀合前体的合成
[0104]
gal(n/o),glu(n/o),man(n/o)缀合前体的合成路线如图3所示。
[0105]
化合物13的合成
[0106]
取d-氨基葡萄糖盐酸盐10g(46.37mmol),4-二甲基氨基吡啶(4-dmap)548mg(4.6mmol),溶于100ml无水dcm中,加入et3n 38ml(278mmol),冰浴条件下,向反应体系中缓慢滴加ac2o 36ml(340mmol)。温度升至室温,搅拌反应24小时。加水淬灭反应,乙酸乙酯萃取。有机层使用饱和nahco3洗涤,饱和nacl洗涤,无水硫酸钠干燥。旋蒸干燥后,乙酸乙酯-石油醚5∶1体系10倍量溶剂重结晶,析出白色针状结晶。抽滤,用少量乙酸乙酯-石油醚1∶1洗涤,干燥,得9.37g(24.06mmol)白色棉絮状固体化合物13,产率51.92%。1h nmr(400mhz,cdcl3)δ6.16(d,j=3.7hz,1h),5.70(d,j=9.0hz,1h),5.30-5.13(m,2h),4.53-4.41(m,1h),4.24(dd,j=12.5,4.1hz,1h),4.06(dd,j=12.5,2.4hz,1h),4.02-3.94(m,1h),2.18(s,3h),2.07(s,3h),2.04(s,3h),2.03(s,3h),1.93(s,3h).esi-ms for c
16h23
no
10
[m+h]
+
found 390.1420,calcd 390.1395;[m+na]
+
found412.1241,calcd 412.1215.
[0107]
化合物14的合成
[0108]
称取化合物1310g(25.7mmol)溶于100ml无水dcm中,氩气保护,冰浴条件下缓慢滴加三氟甲磺酸三甲基硅酯10ml(51.7mmol),30℃反应3小时,冰浴下滴加n,n-二异丙基乙胺(dipea)18.0ml(103.4mmol),搅拌20min。蒸干溶剂柱层析分离,ea∶pe=2∶1。分离得透明油状物化合物146.7g(20.3mmol),产率79.0%。1h nmr(400mhz,dmso)δ6.03(d,j=7.3hz,1h),5.06(t,j=2.4hz,1h),4.79(dt,j=9.4,1.6hz,1h),4.15-4.06(m,3h),3.61-3.55(m,1h),2.06(s,3h),2.04-2.02(m,6h),2.00(d,j=1.9hz,3h).esi-ms for c
14h19
no8[m+h]
+
found 330.1207,calcd 330.1184.esi-ms for c
14h19
no8[m+h]
+
found 330.1257,calcd 330.1184.
[0109]
化合物16的合成
[0110]
称取化合物15(7.6g,64.7mmol),化合物7(10g,29.4mmol)溶解于300ml dcm中,加入10ml(73.5mmol)三乙胺,室温搅拌10h。过滤除去溶剂,滤饼用dcm洗涤多次,得白色粉末化合物169.3g(26.9mmol),产率91.8%。1h nmr(400mhz,dmso)δ7.71(t,j=5.6hz,2h),4.33(s,2h),3.40-3.35(m,4h),3.04-2.95(m,4h),2.09-1.95(m,4h),1.46-1.33(m,12h),1.29-1.22(m,8h).esi-ms for c18h
36
n2o4[m+h]
+
found 345.2894,calcd 345.2753;[m+na]
+
found 367.2724,calcd367.2573.
[0111]
化合物17的合成
[0112]
称取化合物3(500mg,1.52mmol)溶解于15ml三氯乙烷(dce)中待用,称取化合物16(626mg,1.82mmol)溶解于5ml dce中,加入tmsotf 0.33ml(1.82mmol),搅拌下缓慢滴加dce溶解的化合物3,35℃反应24h。后将反应液用饱和nahco3淬灭,饱和nacl洗涤,无水硫酸钠干燥。dcm∶meoh=20∶1柱层析分离,得白色固体化合物17216mg(0.32mmol),产率23.4%。1h nmr(400mhz,dmso)δ7.83(d,j=9.2hz,1h),7.76-7.69(m,2h),5.22(d,j=3.4hz,1h),4.97
nmr(400mhz,dmso)δ7.72(td,j=5.7,2.6hz,2h),5.13-5.05(m,3h),4.86(s,1h),4.32(t,j=5.0hz,1h),4.15(dd,j=12.2,5.4hz,1h),4.05(dd,j=12.2,2.6hz,1h),3.95-3.88(m,1h),3.62(dt,j=9.7,6.8hz,1h),3.45(dt,j=9.7,6.4hz,1h),3.41-3.31(m,3h),3.04-2.98(m,4h),2.11(s,3h),2.05-2.01(m,10h),1.94(s,3h),1.60-1.53(m,2h),1.45-1.24(m,18h).esi-ms for c
32h54
n2o
13
[m+h]
+
found 675.3795,calcd675.3704;[m+na]
+
found 697.3616,calcd 697.3524
[0121]
化合物24的合成
[0122]
称取化合物22(300mg,0.77mmol)溶解于15ml dce中,称取化合物19(435mg,1.54mmol)溶解于5ml dce中,加入bf3et2o 0.6ml(4.61mmol)溶解待用,搅拌下缓慢滴加dce溶解的化合物22,35℃反应48h。后将反应液用饱和nahco3淬灭,水洗除去六甘醇原料,饱和nacl洗涤,无水硫酸钠干燥。dcm∶meoh=20∶1柱层析分离,得白色固体化合物24 132mg(0.22mmol),产率28.0%。1h nmr(400mhz,dmso)δ5.13-5.10(m,3h),4.92(d,j=1.4hz,1h),4.16(dd,j=12.2,5.1hz,1h),4.10(d,j=4.9hz,1h),4.05(dd,j=12.3,2.7hz,1h),4.03-3.98(m,1h),3.53-3.51(m,24h),2.12(s,3h),2.03-2.02(m,6h),1.95(s,3h).esi-ms for c
26h44o16
[m+nh4]
+
found 630.2969,calcd 630.2973.
[0123]
1.2.3 gal/glu/man缀合前体活泼酯的合成
[0124]
gal/glu/man缀合前体活泼酯的合成路线如图4所示。
[0125]
化合物25-1的合成
[0126]
将化合物3(400mg,1.22mmol),5-己烯-1-醇(158.2mg,1.58mmol)溶解于8ml无水dce中,冰浴下缓慢滴加tmsotf(216.1mg,0.97mmol)。室温反应5小时,加入2eq dipea,搅拌10min。蒸干溶剂,柱层析分离,ea∶pe=2∶1。得产物451mg(1.05mmol),产率86.1%。1h nmr(400mhz,dmso)δ7.82(d,j=9.2hz,1h),5.85-5.76(m,1h),5.22(d,j=3.4hz,1h),5.03-4.93(m,3h),4.49(d,j=8.5hz,1h),4.05-4.01(m,3h),3.87(dt,j=11.3,8.8hz,1h),3.72(dt,j=9.9,6.1hz,1h),3.43(dt,j=10.0,6.5hz,1h),2.11(s,3h),2.07-2.01(m,2h),2.00(s,3h),1.90(s,3h),1.77(s,3h),1.51-1.44(m,2h),1.41-1.34(m,2h).esi-ms for c
20h31
no9[m+h]
+
found 430.2082 calcd 430.2077;[m+na]
+
found 452.1906,calcd452.1987.
[0127]
化合物26-1的合成
[0128]
将化合物14(400mg,1.22mmol),5-己烯-1-醇(158.2mg,1.58mmol)溶解于8ml无水dce中,冰浴下缓慢滴加tmsotf(216.1mg,0.97mmol)。室温反应5小时,加入2eq dipea,搅拌10min。蒸干溶剂,柱层析分离,ea∶pe=2∶1。得产物396mg(0.92mmol),产率75.7%。1h nmr(400mhz,dmso)δ7.94(d,j=9.1hz,1h),5.84-5.73(m,1h),5.11-5.05(m,1h),5.00(dq,j=17.2,1.7hz,1h),4.94(ddt,j=10.2,2.3,1.2hz,1h),4.82(t,j=9.7hz,1h),4.59(d,j=8.5hz,1h),4.18(dd,j=12.3,4.8hz,1h),4.01(dd,j=12.3,2.4hz,1h),3.82(ddd,j=10.0,4.8,2.5hz,1h),3.74-3.66(m,2h),3.44(dt,j=9.9,6.4hz,1h),2.05-1.99(m,5h),1.97(s,3h),1.91(s,3h),1.75(s,3h),1.53-1.45(m,2h),1.41-1.31(m,2h).esi-ms for c
20h31
no9[m+h]
+
found 430.2082 calcd 430.2077;[m+na]
+
found452.1906,calcd 452.1987.
[0129]
化合物27-1的合成
h]-found 447.1504calcd 447.1508.
[0137]
化合物25的合成
[0138]
化合物25-2(200mg,0.45mmol),edci(172.5mg,0.90mmol),nhs(103.6mg,0.90mmol)溶解于5ml无水dcm中,氩气保护下反应5小时。饱和nahco3洗涤2次,饱和食盐水洗涤1次,无水na2so4干燥,旋蒸干燥,得产物150mg(0.28mmol),产率63.2%。1h nmr(400mhz,dmso)δ7.83(d,j=9.2hz,1h),5.22(d,j=3.4hz,1h),4.96(dd,j=11.2,3.4hz,1h),4.51(d,j=8.5hz,1h),4.09-3.99(m,3h),3.90(dt,j=11.2,8.7hz,1h),3.75(dt,j=10.6,5.5hz,1h),3.46(dt,j=9.9,5.9hz,1h),2.82(s,4h),2.68(t,j=7.3hz,2h),2.11(s,3h),2.00(s,3h),1.90(s,3h),1.77(s,3h),1.68-1.54(m,4h).esi-ms for c
23h32
n2o
13
[m+nh4]
+
found545.1973 calcd 545.1983;[m+na]
+
found 567.1794,calcd 567.1802.
[0139]
化合物26的合成
[0140]
化合物26-2(200mg,0.45mmol),edci(172.5mg,0.90mmol),nhs(103.6mg,0.90mmol)溶解于5ml无水dcm中,氩气保护下反应5小时。饱和nahco3洗涤2次,饱和食盐水洗涤1次,无水na2so4干燥,旋蒸干燥,得产物142mg(0.27mmol),产率59.9%。1h nmr(400mhz,dmso)δ7.95(d,j=9.2hz,1h),5.10-5.04(m,1h),4.83(t,j=9.7hz,1h),4.60(d,j=8.5hz,1h),4.18(dd,j=12.3,4.8hz,1h),4.02(dd,j=12.3,2.5hz,1h),3.83(ddd,j=10.0,4.8,2.5hz,1h),3.77-3.68(m,2h),3.48(dt,j=10.0,5.9hz,1h),2.81(s,4h),2.68(t,j=7.2hz,2h),2.02(s,3h),1.97(s,3h),1.91(s,3h),1.75(s,3h),1.70-1.56(m,4h).esi-ms for c
23h32
n2o
13
[m+nh4]
+
found 545.1974 calcd 545.1983;[m+na]
+
found567.1797,calcd 567.1802.
[0141]
化合物27的合成
[0142]
化合物27-2(200mg,0.45mmol),edci(172.5mg,0.90mmol),nhs(103.6mg,0.90mmol)溶解于5ml无水dcm中,氩气保护下反应5小时。饱和nahco3洗涤2次,饱和食盐水洗涤1次,无水na2so4干燥,旋蒸干燥,得产物120mg(0.22mmol),产率48.9%。1h nmr(400mhz,dmso)δ5.16-5.04(m,3h),4.88(d,j=1.5hz,1h),4.15(dd,j=12.2,5.3hz,1h),4.05(dd,j=12.2,2.6hz,1h),3.97-3.89(m,1h),3.70-3.63(m,1h),3.53-3.48(m,1h),2.82(s,4h),2.76-2.69(m,2h),2.11(s,3h),2.05-1.99(m,6h),1.94(s,3h),1.76-1.62(m,4h).esi-ms for c
23h31
no
14
[m+nh4]
+
found 563.2108 calcd 563.2088;[m+na]
+
found 568.1660,calcd 568.1642.
[0143]
第二部分 各核酸缀合物的合成
[0144]
实施例3亚磷酰胺固相合成法
[0145]
对于已是亚磷酰胺单体的缀合结构(如sg),在制备得到纯品后,用无水乙腈将其配置成0.05g/ml的溶液,采用一般固相合成方法使用寡核苷酸合成仪可程序化自动缀合于寡核苷酸(on)5
′
末端,合成完毕后遵循核酸链标准流程进行分离纯化可获得sg-on缀合物,总收率大致在20%左右。针对化合物17,18,20,21,23,24,需额外增加制备亚磷酰胺前体的过程。其制备过程可简单描述为在无水乙腈,氩气保护下,与1.5倍当量的2-氰乙基-n,n,n
′
,n
′‑
四异丙基亚磷酰二胺和1h-四氮唑反应3h,蒸干溶剂后用dcm复溶,饱和nahco3洗涤2次,饱和食盐水洗涤1次,无水na2so4干燥,旋蒸干燥。无需进一步柱层析纯化,按0.05g/ml的浓度用无水乙腈稀释后,直接用于核酸合成。固相合成完毕后经进一步的分离纯化,可得到
相应的gal(n/o),glu(n/o),man(n/o)缀合物。
[0146]
实施例4活泼酯液相汇聚法
[0147]
液相溶液汇聚法合成g3-on流程图如图5所示。针对缀合前体是活泼酯结构的化合物(g3ac-nhs,compound 25,26,27),以g3ac-nhs为例(图5),首先在合成完目标序列的cpg基础上,利用固相合成仪合成一段带氨基末端的linker1,得到中间体1a,用于后续连接。然后将此中间体用浓氨水60℃切割12小时,反应完毕后取上清,cpg粉末用纯水洗涤,液体浓缩除氨后,使用gilson制备型hplc液相分离。梯度洗脱方法:acn/0.05m teab;0min-5%acn,22min-22%acn;4ml/min;xbridge oligonucleotide beh c18 obd prep column,2.5μm,30mm x 50mm;得到带裸露氨基末端的核酸中间体1b(收率30%),收集馏分冻干后在1m teab缓冲液的条件下与5倍量g3ac-nhs活泼酯反应5小时得到中间体1c(单步收率92%),反应液旋蒸干燥后用浓氨水60℃切割12小时,后除氨再次液相分离(分离条件同前)。收集馏分浓缩后,使用葡聚糖凝胶柱(ge hiprep
tm 26/10 desalting)脱盐,得纯品g3-on(总收率约22%,maldi-tof ms分析确证),定量后冻干备用。
[0148]
实施例5 galnac缀合荧光标记物的合成
[0149]
溶液汇聚法合成cy5.5-g3-on荧光标记物流程图如图6所示。此法更换了氨基linker2的结构并且使用了3
′
nh cpg,同样在将此linker程序化连接于核酸链末端后,在dmt-off的情况下,中间体2a暴露出氨基末端,直接将此cpg粉末与5倍量的g3ac-nhs活泼酯在1m teab的溶液环境下反应5小时得中间体2b,然后使用上述同样方法氨切及液相分离方法得到暴露出3
′
nh的中间体2c(收率25%),中间体2c冻干后与5倍量cy 5.5活泼酯在ph 8的硼酸盐缓冲液体系下反应4小时得最终产物,偶联产物再经一次液相纯化(梯度洗脱方法:acn/0.05m teab;0min-10%acn,30min-80%acn;4ml/min)得目标物纯品cy5.5-g3-on荧光标记物(总收率20%),冻干备用即可。
[0150]
第三部分 反义核酸缀合物及其制剂的基本性质及体内外药效
[0151]
实施例6反义核酸ct102、ct102
moe5
及其不同缀合物的血清稳定性
[0152]
反义核酸为ct102
moe5
的序列为:tesmces
mm
cestesmcesmcsgsgsasgsmcsmcsasgsasmcestestes m
cesae;
[0153]
其中:mc表示5-甲基胞嘧啶修饰(5mc);s表示磷硫代修饰(ps);e表示2
′‑
o-moe修饰(2
′‑
o-moe);结构分别如下所示:
[0154][0155]
反义核酸糖基缀合物选自以下化合物:
[0156][0157]
材料与方法:
[0158]
50%的胎牛血清孵育样品,用适当量程移液枪分别取16μl aso或其缀合物(50μm)+200μl fbs+184μl pbs至600μl ep管中,置于37℃恒温水浴中,在实验设计时间点(0h,4h,8h,12h,......)取出10μl样品,涡旋离心后立即放入-80℃保存。收集完毕所有时间点样品后进行凝胶电泳分析:20%聚丙烯酰胺凝胶电泳分析aso血清代谢稳定性。page胶配制方法如下:
[0159]
表1. 20%聚丙烯酰胺凝胶电泳试剂比例
[0160][0161]
上样:向每个样品管中加入2μl 6
×
dna loading buffer,混匀离心后上样,如沉降效果不好可适当增加loading buffer的量。电泳:110v电压分离。通常情况下蓝色条带移至另一端边缘时(2-2.5h)即可切断电源,将凝胶取下。染色:将胶放入20ml 1
×
tbe的溶液中加入5μl sybr gold染料染色10min。成像:使用高灵敏化学发光凝胶成像分析系统(chemidoc xrs system)成像,结果使用image lab处理分析。注意:染色及照相过程需避光处理。
[0162]
结果:总体来说,原始序列ct102因具有全硫代化学修饰,其耐酶解能力及血清稳定性已经相对优秀,8天长时间血清中未完全降解(图7)。而在此基础上的修饰物稳定性更加突出,核酸酶抗性进一步增强,在实验中的所有时间点降解甚少。修饰物已经具备了很强的血清稳定性,在此基础上的缀合物对血清稳定性的增益不明显。同时,在本实验发现了一个特殊现象是g3-ct102
moe5
降解明显,在原始链下方会出现明显的条带,通过加入ct102
moe5
对比发现此条带就是ct102
moe5
,提示g3-ct102
moe5
在血清中会逐渐脱落g3缀合结构,暴露出ct102
moe5
继续发挥活性。而其余缀合物未发现此种现象,这可能是与缀合基团的大小相关的,结构较大的基团容易被酶识别降解。
[0163]
实施例7反义核酸ct102及其缀合物制剂给药后在肝癌细胞中转染入胞能力
[0164]
材料与方法:
[0165]
1、dnca/cld/peg与反义核酸(aso)或其缀合物混合形成脂质复合物的制备:
[0166]
因实验目的的不同,溶液浓度及载体比例有所不同(dnca/cld/peg/aso或其缀合物=30∶30∶0.6∶1、20∶20∶0.4∶1、40∶20∶0.6∶1或20∶40∶0.6∶1),此处仅以制备体内分布实验所需脂质复合物为例(dnca/cld/peg/aso或其缀合物=30∶30∶0.6∶1),其余实验除浓度及组分配比不同外,方法相同。向无菌无酶的ep管中加入100μl genopti溶液,随后依次加入10μl浓度为200μm的aso,1.2μl浓度为50mm的dnca溶液和1.2μl浓度为50mm的cld溶液,再加入0.6μl浓度为2mm的dspe-peg溶液,补足剩余genopti溶液至200μl,50℃超声20min即可使用。
[0167]
所述dnca、cld、dspe-peg结构式如下所示:
[0168][0169]
cy5.5标记的具体结构有如下三种:
[0170][0171]
2.应用流式细胞术考察包载各核酸样本(cy5.5标记)在人肝癌细胞hepg2、huh7中跨膜转染能力
[0172]
将hepg2/huh7细胞分别按100000个/孔、50000个/孔铺至12孔板,37℃培养24h后进行转染,其中核酸浓度为100nm,每孔溶液体积为100μl。给药4小时后,吸去上清液,将待测细胞用pbs洗涤1次,在每孔中加入200μl 0.25%胰蛋白酶,消化2min后,每孔加600μl 10%dmem培养基,将细胞消化下来转移到离心管,1000rpm 3min离心去上清液。用pbs洗涤2次,每份样品加入100μl 10%dmem培养基使细胞重悬,过筛,采用facs caiibur流式细胞分析仪(becton dickinson,sanjose,ca,usa)进行检测。
[0173]
结果:4h后(图8),在hepg2细胞中,药物摄取率分别为cy5.5-ct102(64.4%),cy5.5-ct102
moe5
(70.4%),cy5.5-g3-ct102
moe5
(74.7%),cy5.5-glu-ct102
moe5
(77.8%),在huh7细胞中,药物摄取率分别为cy5.5-ct102(85.0%),cy5.5-ct102
moe5
(88.6%),cy5.5-g3-ct102
moe5
(89.3%),cy5.5-glu-ct102
moe5
(92.6%)。因此,galnac缀合物及glu缀合物均能在一定程度上提高细胞对药物的摄取,推测可能是细胞表面存在糖基受体导致药物被更多的介导内吞导致的。此外,摄取率的提高在hepg2细胞中更加明显。
devices,california,usa)检测450nm处的吸光度,按照如下公式进行计算细胞生存率:
[0185]
cell viability=(r
a-re)/(r
b-re)
×
100%
[0186]
注:ra、rb、re分别代表实验组,无转染试剂组以及空白对照组的吸光度。
[0187]
靶基因沉默活性:将hepg2、huh7细胞分别按100000个/孔、50000个/孔铺至12孔板,37℃培养24h后进行转染。实验组按实施例5制剂方法配置各样本,其中核酸浓度为100nm,溶液体积为200μl,各组分比例为dnca/cld/peg/aso=20∶20∶0.4∶1,进行总rna提取纯化及逆转录检测。
[0188]
细胞总rna的提取和纯化:转染后48h后用trizol法提取total rna。12孔板按500μl/孔的量加入trizol混匀,室温静置5min后在4℃条件下13000rpm离心10min取上清。再加入0.2ml氯仿,摇匀后室温静置15min,溶液自然分为三层。在4℃条件下,13000rpm,离心10min,取上层水相。再加入0.5ml异丙醇,蜗旋后室温静置15min,在4℃条件下13000rpm离心10min,弃上清,管底可见rna沉淀。之后加入1ml 70%乙醇,轻轻洗涤沉淀,4℃条件下10000rpm离心8min弃上清,室温放置10min,晾沉淀至近干。加入depc水溶解,-80℃保存。
[0189]
rna逆转录及检测:总rna的加入量为500ng,无酶水补足5μl后,放入pcr仪,70℃,10min;按照试剂盒说明书配置各组分。pcr执行程序42℃,15min,95℃,5min;4℃保存。随后将10μl上述cdna用40μl无酶水稀释5倍,按照标准程序进行实时定量pcr(40个循环)。其中igf1r上下游引物分别为(5
′‑3′
):atc gtt cat cca agg ctg ttac,agc aat gag acc tgt gtg cctg。内参(β-actin)上下游引物分别为(5
′‑3′
):cca acc gcg aga aga tga,cca gag gcg tac agg gat ag。
[0190]
结果:通过细胞增殖抑制及靶标沉默活性考察比较各缀合物的活性差异,从总体结果看(图12),各缀合物不会影响序列的反义活性,部分组别活性稍优;靶基因沉默水平上看,各缀合物组别未见明显差异,gal-ct102
moe5
,glu-ct102
moe5
,man-ct102
moe5
活性略优。cck8结果显示,各缀合物普遍在hepg2细胞上的活性较好,这应该是hepg2细胞上较huh7更多的表达糖基受体导致的。另外cck8结果发现,单尾缀合物linker的长度不宜过长,否则会影响活性,长链缀合结构(gal(n/o),glu(n/o),man(n/o))较短链缀合结构(gal,glu,man)活性稍差。综合以上结果,在体外确定了gal-ct102
moe5
,glu-ct102
moe5
,man-ct102
moe5
,g3-ct102
moe5
为最优结构,进行了后续的动物水平体内药效实验。
[0191]
实施例11 ct102在多种给药方案下的体内药效
[0192]
材料与方法:
[0193]
以荧光素酶基因标记的人肝癌细胞(hepg2-luc)在小鼠腋下(200w/只)成瘤后将肿瘤组织块均匀分割通过手术移植至小鼠体内肝脏制成模型。整个实验周期为28天,以索拉非尼(每天灌胃给药,连续给药20天)为阳性对照,考察ct102修饰物、缀合物包载组(每4日尾静脉注射给药,共给药6次)的抗肝癌药效。设置生理盐水组(blank)、索拉非尼为阳性对照组,dnca/cld/dspe-peg2000(mix-20/20/0.4/1)包载ct102、其2
′‑
moe修饰物组、ct102
moe5
四种不同的糖基缀合物组。首次给药后每7天对各组小鼠腹腔注射底物luciferin后进行活体成像检测,观察并比对各组小鼠的肿瘤进展情况,28天后处死小鼠,取肝脏拍照、称重(依情况决定是否将肿瘤块从肝脏中剥离),分析比对各组药物对肿瘤生长的作用,制作肝脏冷冻切片,对细胞内igf-ir蛋白进行免疫荧光染色并读片,观察靶标表达抑制情况,制作肝脏石蜡切片,对肝脏组织进行病理分析;同时取小鼠外周血,分离血浆,进行可表
征肝肾功能的各项血生化检测,结合整个实验周期中小鼠的体重情况分析药物安全性(表2)。
[0194]
表2.抗肝癌aso药物制剂实验方案
[0195][0196]
blank(saline)及反义核酸制剂组通过尾静脉给药6次、sorafenib灌胃给药20次实验结果(图13)表明:dnca/cld/dspe-peg2000包载的ct102
moe5
及各类缀合物的肿瘤抑制效果显著优于ct102及索拉非尼,表明反义核酸ct102经过2
′‑
moe能够增强肿瘤增长抑制活性;其中,ct102
moe5
各缀合物组(g3/gal/glu/man)均优于非缀合的ct102
moe5
组,d28天肿瘤平均大小分别为d0天的15,9,7,18倍(ct102
moe5
24倍)。其中缀合物gal及glu活性最强,对肝癌肿瘤抑制效果显著优于ct102(p<0.05),为抗肝癌的优秀候选序列。
[0197]
第四部分 抗肝癌反义核酸新序列设计及其在抗肝癌中的应用
[0198]
实施例12反义核酸的来源和设计
[0199]
参考ct102,以igf1r mrna为靶标设计得到一系列抗肝癌反义核酸新序列(表3)。首先,靶向外显子区域最有可能得到有效的反义核酸。其次有效地结合靶mrna,还取决于aso长度、序列内容、二级结构、热力学性质和靶标可及性等。通常将20nt作为最佳长度,同时序列中gc的百分比最好不低于40%,tm值不低于48℃均为有效aso的设计原则(表4)。根据文献已报道的具有抗肝癌活性的序列作为候选序列,如phn01、phn02、phn03序列(来自文献[progbiochem biophys,2002,29(2):247-251.]),与ct102一样,phn01、phn02及phn03序列均靶向igf1r mrna序列中蛋白编码区(coding sequence,cds)第一段。同时通过gc比、tm值及二级结构模拟等反义核酸设计原则新设计靶向igf1r mrna蛋白编码区第一段的序列phn04和phn05,以及靶向cds第二段区域的phn06和phn07。
[0200]
表3
[0201][0202]
表4
[0203][0204][0205]
实施例13各反义核酸新序列在hcc细胞增殖抑制、靶基因沉默及细胞凋亡方面与ct102的差异
[0206]
材料与方法:
[0207]
各反义核酸新序列(表5所示)订购自生工生物工程(上海)股份有限公司。
[0208]
表5
[0209][0210]
细胞增殖抑制、靶基因沉默活性实验与实施例10一致。
[0211]
流式细胞仪检测细胞凋亡:hepg2细胞在12孔板中以1
×
105细胞/孔的密度培养18小时后,将细胞暴露于上述反义核酸制剂。继续孵育24h,弃去培养基,然后根据制造商的方案将细胞胰蛋白酶消化,收集并用膜联蛋白annexin v-fitc/pi细胞凋亡检测试剂盒染色,通过流式细胞术分析凋亡细胞的比例。
[0212]
结果:cck-8实验中,针对两种不同类型肝癌细胞(hepg2及huh7),新序列phn01、phn02、phn03、phn07均表现出优于ct102的抗细胞增殖效果(图14a)。rt-qpcr实验中,phn02、phn03、phn04、phn06、phn07均表现出优于ct102的igf1r mrna沉默效果(图14b)。细胞凋亡实验中,phn02及phn07促肝癌细胞晚期凋亡的效果优于ct102,而早期凋亡状态可逆,晚期凋亡状态不可逆,说明phn02抗肝癌效果优于ct102(图15)。
[0213]
实施例14反义核酸新序列phn02和phn07在a549、mcf-7、b-cpap细胞增殖抑制、靶基因沉默活性
[0214]
材料与方法:
[0215]
cck-8检测法:将a549、mcf-7、b-cpap细胞分别按3000个/孔、3000个/孔、6000个/孔铺至96孔板,37℃培养24h后进行转染,方法同实施例10;
[0216]
靶基因沉默活性:将a549、mcf-7、b-cpap细胞分别按50000个/孔、50000个/孔、60000个/孔铺至12孔板,37℃培养24h后进行转染,方法同实施例6。
[0217]
结果:phn02和phn07对a549、mcf-7、b-cpap细胞的增值抑制活性同ct102相当,对a549和b-cpap有较好的增殖抑制作用;phn02和phn07对a549、mcf-7、b-cpap细胞中igf1r mrna的沉默效果与ct102持平,无显著差异(图16)。
[0218]
实施例15 phn02的2
′‑
o-moe修饰物在细胞增殖抑制、靶基因沉默方面与ct102、phn02的差异
[0219]
材料与方法:
[0220]
对ct102及phn02进一步修饰,得到两端各5个2
′‑
o-moe修饰且所有胞嘧啶为5mc的序列ct102
moe5
及phn02
moe5
。反义核酸修饰物如表6所示,由实验室合成。细胞增殖抑制活性及
靶基因沉默活性操作同实施例10。
[0221]
表6
[0222][0223]
结果:结果表明(图17)phn02
moe5
与ct102
moe5
抗肝癌效果基本持平,phn02
moe5
、ct102
moe
抗肝癌效果比phn02、ct102进一步提高,phn02
moe5
药效较对hepg2和huh-7细胞的增殖抑制作用和igf1r mrna沉默效果较ct102均有显著提高。
[0224]
实施例16 mix包载反义核酸phn02
moe5
对原位组织移植瘤肝癌小鼠的药效情况
[0225]
材料与方法:
[0226]
方法同实施例11,具体实验方案如下表7所示:
[0227]
表7
[0228][0229]
结果:ct102和phn02的2
′‑
o-moe修饰物ct102
moe5
及phn02
moe5
显示出优于4倍剂量裸给ct102的药效,且新序列phn02
moe5
与ct102
moe5
药效相当(图18b、c);通过rt-qpcr对肿瘤中igf1r的mrna表达水平的检测显示修饰物ct102
moe5
及phn02
moe5
较4倍量裸给ct102更显著降低肿瘤中igf1r mrna的表达(p<0.01)(图18d)。整个实验周期小鼠状态良好,体重正常(图18e);小鼠肿瘤成像图观察到各时间点肿瘤的变化情况,包载组ct102
moe
(c组)和phn02
moe5
(d组)的肿瘤增长较生理盐水组和4倍量裸给ct102(b组)相比,肿瘤增长缓慢(图19)。
[0230]
本文显示并详细描述的信息足以实现本发明的上述目的,因此本发明的优选实施方案代表本发明的主题,该主题为本发明所广泛涵盖。本发明的范围完全涵盖其它对本领域技术人员来说显而易见的实施方案,因此,本发明的范围不被除所附权利要求之外的任何内容所限制,其中除了明确说明外,所用元素的单数形式并不是指“一个和唯一”,而是指“一个或更多”。对本领域一般技术人员来说,所有公知的上述优选的实施方案和附加实施方案部分的结构、组成和功能上的等价物因此引入本文作参考,而且试图被本发明的权利要求所涵盖。
[0231]
此外,不需要某种设备或方法来表达本发明所解决的每个问题,因为它们都已包括在本发明的权利要求之内。另外,无论本发明公开事实中的所有部分、成分,或者方法步骤是否在权利要求中被明确叙述,它们都没有贡献给公众。但是,对本领域普通技术人员来
说,很明显在不背离如所附权利要求中所阐明的本发明的实质和范围的前提下,可以在形式、试剂和合成细节上做出各种改变和修饰。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1