基因标志物在恶性肺结节筛查中的应用、筛查模型的构建方法和检测装置与流程
1.本发明涉及对影像表现为高风险的肺结节(radiographically highly suspicious lung nodules)良恶性早筛, 属于分子生物医学技术领域。
背景技术:
2.肺癌是其中一个世界上确证率最高的癌症,高风险人群包括65岁以上,具有下列一项以上的危险因素者。危险因素包括:重度吸烟,曾经拥有抽烟史,家庭病史,接受过胸腔放射性治疗,致癌物质。由于肺癌前期缺乏明显症状,患者一般于肺癌中后期(stage iii, iv)被诊断出。然而,大量研究表明在早期被诊断出的肺癌患者能有更高的生存率。在肺癌一期(stage i)被诊断出的患者比在肺癌四期(stage iv)被诊断出的患者的五年存活率提高13倍。因此,肺部肿瘤早发现和早诊断对于提高肺癌患者的生存率至关重要。
3.低剂量胸腔电脑断层扫描(low-dose computed tomography thorax, ldct)检测肺结节是现今发现肺部肿瘤最常见的诊断方式。针对影像学判定的肺结节进行手术切除,能有效地减少20%-39% 的肺癌死亡率。然而,大约15%-35% 的肺结节,在初始ldct影像表现判定为高风险的肺结节,在手术切除后,最后被确认为病理性无害。因此,影像学检测存在一定的局限性,仅依据影像检测结果进行恶性肺肿瘤诊断,增加了一些非必要的手术,造成患者非必要的手术风险和并发症风险,和增加了医疗支出负担。因此,对仅在影像学依据下判定为肺癌高危人群的肺结节良恶性判断工作尤为重要。
技术实现要素:
4.本发明所要解决的技术问题是:现有技术中针对肺癌结节良恶性进行诊断过程中缺少无创方式的检测手段,导致了非必要的手术、增加了患者负担。
5.本专利的技术方案中,提供了一种对血浆样本cfdna进行wgs测序,通过对高通量测序结果,获取片段信息,进行对良恶性肺结节差异dna片段长度比值(fragment size ratio), 读段5端断裂点序列读段占比(breakpoint motif), 1mb 窗口拷贝数变化(1 mb-bin copy number variant), 16bp肿瘤新短序列(16bp neomers)和核小体覆盖模式 (nucleosome coverage patterns),利用弹性网络逻辑回归模型(elastic net logistic regression, glm), 极致梯度提升(extreme gradient boosting,xgboost),随机森林(random forest)和神经网络(neural network)四种算法利用自动化机器学习构建多特征多算法整合模型,实现对恶性肺结节无创精准诊断。
6.具体的技术方案是:基因标志物在制备恶性肺结节筛查试剂中的应用;所述的基因标志物包括:第一标志物:cfdna片段比对至参考基因组的不同窗口中的短读段数量占比和长读段数量占比;
第二标志物:不同种类的cfdna片段比对至参考基因组的5’端的m个碱基片段在全部碱基片段中的占比;第三标志物:wgs数据中染色体上不同窗口中的拷贝数;第四标志物:肿瘤新短序列占比;第五标志物:核小体覆盖模式。
7.所述的第一标志物通过如下步骤获得:将读段数据结果比对至参考基因组,将参考基因组划分为多个窗口,并分别获得在每个窗口范围内的短读段数量和超长读段数量占比。
8.所述的第二标志物通过如下步骤获得:将读段数据中的5’端的m个碱基数据作为碱基片段集合,并得到各种碱基片段在全部片段中所占比例。
9.所述的第三标志物通过如下步骤获得:将参考基因组划分为多个窗口,并分别获得wgs数据在1-22号染色体上不同窗口中的拷贝数数据。
10.所述的第四标志物通过如下步骤获得:穷举法生成长度为16bp的短序列集合a;在人类参考基因序列中穷举出所有的长度为16bp短序列集合b,从集合a中将集合b数据剔除后,定义为无效子;从癌症数据库中获得不同癌种的样本wgs测序结果,提取出多次出现的碱基替换突变; 根据碱基替换的位置,从无效子中找到包含这些碱基替换的无效子短序列集合c;获取东亚人群中频率大于0.01的碱基替换突变;根据碱基替换的位置,从无效子中找到包含这些碱基替换的无效子短序列集合d;从集合c中将集合d的无效子序列排除,定义为新短序列;统计出样本中能够读取到任意一个新短序列的样本数量,再针对每一个新短序列,搜索出包含这些新短序列的样本数量,将每一个新短序列的样本数量与所有能读取到任意新短序列的样本总数的比例。
11.所述的第五标志物通过如下步骤获得:从gtrd数据库中获得转录因子,并从中排除掉不在cis-bp数据库中有已知转录位点的转录因子;将获得的转录因子的转录位点附近-5kb到+5kb范围作为窗口,获得可以比对至这些窗口中的长度为100-220bp的片段,对窗口中的读段数据依次进行gc校正和测序深度平滑处理,得到每个转录因子的覆盖模式曲线;对于每个转录因子,获得如下三个特征,共同作为核小体覆盖模式:1)对于转录因子的全部转录位点,求出转录位点的上端1kb到下端1kb的平均深度;2)对于获得的覆盖模式曲线,获得曲线波谷的幅度值,作为转录因子的中心深度;3)对于获得的覆盖模式曲线进行快速傅里叶变换,获得核小体振幅信号的最高点的振幅数值。
12.恶性肺结节筛查模型的构建方法,包括如下步骤:步骤1,对阳性组和对照组的样本进行cfdna的提取并测序,获得读段数据;步骤2,将读段数据结果比对至参考基因组,将参考基因组划分为多个窗口,并分别获得在每个窗口范围内的短读段数量占比和长读段数量占比,作为第一特征集合;
步骤3,将读段数据中的5’端的m个碱基数据作为碱基片段集合,并得到各种碱基片段在全部片段中所占比例作为第二特征集合;步骤4,将参考基因组划分为多个窗口,并分别获得在每个窗口范围内的拷贝数数据,作为第三特征集合;步骤5,将读取到16bp新短序列的的样本数与所有能读取到任意新短序列的样本总数的比例,作为第四特征集合;步骤6,所选取的转录因子的核小体覆盖模式特征,作为第五特征集合;步骤7,以第一、第二、第三、第四和第五特征集合共同作为初始特征值,作为模型特征向量输入至分类模型中,并以肺结节良恶性作为输出值,对模型进行训练,获得早筛模型。
13.所述的步骤3中包括:步骤3-1,将参考基因组划分为多个窗口,并分别获得每个窗口范围内的长读段数量和短读段数量;步骤3-2,将步骤3-1的所有窗口的短读段数量和长读段数量标准化处理,标准化后的短读段数和长读段数的比例作为第一特征值。
14.所述的步骤3-1中窗口大小是5mb,共划分出541个窗口。
15.所述的短读段是指长度100-150bp,所述的长读段是151-220bp。
16.所述的步骤3中,m是4。
17.所述的步骤4中窗口大小是1mb,共划分出2475个窗口。
18.所述的步骤5 中,作为第四特征集合的获取步骤如下:步骤5-1,穷举法生成长度为16bp的短序列集合a;在人类参考基因序列中穷举出所有的长度为16bp短序列集合b,从集合a中将集合b数据剔除后,定义为无效子;步骤5-2,从癌症数据库中获得不同癌种的样本wgs测序结果,提取出多次出现的碱基替换突变; 根据碱基替换的位置,从无效子中找到包含这些碱基替换的无效子短序列集合c;步骤5-3,获取东亚人群中频率大于0.01的碱基替换突变;根据碱基替换的位置,从无效子中找到包含这些碱基替换的无效子短序列集合d;从集合c中将集合d的无效子序列排除,定义为新短序列;步骤5-4,统计出样本中能够读取到任意一个新短序列的样本数量,再针对每一个新短序列,搜索出包含这些新短序列的样本数量,将每一个新短序列的样本数量与所有能读取到任意新短序列的样本总数的比例,作为模型的第四特征集合。
19.所述的癌症数据库是pcawg数据库。
20.不同癌种是指肠癌、肺癌、乳腺癌、胃癌、前列腺癌和肝癌。
21.东亚人群中的碱基替换突变是通过gnomad数据库获得。
22.所述的步骤6包括:步骤6-1,从gtrd数据库中获得转录因子,并从中排除掉不在cis-bp数据库中有已知转录位点的转录因子;步骤6-2,将步骤6-1中获得的转录因子的转录位点附近-5kb到+5kb范围作为窗口,获得可以比对至这些窗口中的长度为100-220bp的片段,对窗口中的读段数据依次进行
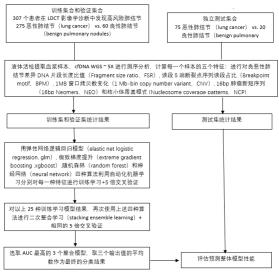
gc校正和测序深度平滑处理,得到每个转录因子的覆盖模式曲线;步骤6-3,对于每个转录因子,获得如下三个特征,共同作为核小体覆盖模式特征:1)对于转录因子的全部转录位点,求出这些转录位点的上端1kb到下端1kb的平均深度;2)对于获得的覆盖模式曲线,获得曲线波谷的幅度值,作为转录因子的中心深度;3)对于获得的覆盖模式曲线进行快速傅里叶变换,获得核小体振幅信号的最高点的振幅数值。
23.所述的步骤7中,分类模型的步骤包括:步骤7-1,分别将第一、第二、第三、第四和第五特征集合输入至不同的分类器模型中,进行模型的训练,并获得分别针对第一、第二、第三、第四和第五特征集合的最优的一个或多个分类器模型;步骤7-2,将步骤7-1中获得的第一、第二、第三、第四和第五特征集合的最优分类器模型进行二次集合训练,构建出集成分类器模型。
24.所述的不同的分类器模型选自弹性网络回归模型(elasticnetlogisticregression,glm)、极致梯度提升(extremegradientboosting,xgboost)、随机森林(randomforest)、深度学习神经网络模型(deeplearning,nn)。
25.二次集合训练中采用广义线性模型(generalizedlinearmodel,glm),极致梯度提升xgboost模型或者深度学习回归模型。
26.一种恶性肺结节检测装置,包括:测序模块,用于对阳性组和对照组的样本进行cfdna的提取并测序,获得读段数据,并且获得wgs测序数据;第一特征获取模块,用于将读段数据结果比对至参考基因组,将参考基因组划分为多个窗口,并分别获得在每个窗口范围内的短读段数量和超长读段数量占比,作为第一特征集合;第二特征获取模块,用于将读段数据中的5’端的m个碱基数据作为碱基片段集合,并得到各种碱基片段在全部片段中所占比例作为第二特征集合;第三特征获取模块,用于将参考基因组划分为多个窗口,并分别获得wgs数据在染色体上不同窗口中的拷贝数数据,作为第三特征集合;第四特征获取模块,将读取到16bp新短序列的的样本数与所有能读取到任意新短序列的样本总数的比例,作为第四特征集合;第五特征获取模块,分析所选取的转录因子的核小体覆盖模式特征,作为第五特征集合;预测模块,用于以第一、第二、第三、第四和第五特征集合共同作为初始特征值,作为模型特征向量输入至分类模型中,并以肺结节良恶性作为输出值,对模型进行训练,获得早筛模型。
附图说明
27.图1是模型构建过程示意图;图2是各个特征中最大贡献值特征的差异分布图;
图3是在训练集单独使用各个特征模型及使用所有特征的模型的auc曲线;图4是在验证集上使用所有特征的模型的auc曲线;图5是是训练集上全部模型集合后分类器的预测分数分布图;图6是验证集上全部模型集合后分类器的预测分数分布图。
具体实施方式
28.本发明中的计算方法详述如下:本发明首先需要进行从血液样品中对cfdna的提取、建库、测序等步骤。这里的提取、建库方法没有特别的限定,可以从现有技术中的提取方法中进行调整。这里的测序过程中可以采用现有技术中的测序技术获得cfdna的碱基信息。
29.本专利中的模型的目的是用于区分区分良性肺结节(benign lungnodules)和恶性肺结节(malignant lungnodules)。对样本进行分类,对在ldct影像检测中判断为高风险的肺结节患者,根据后续术后病理判定为良性肺结节的患者作为对照组,判定为恶性肺结节的患者作为阳性组。
30.本发明中的模型构建过程采用的数据集情况如下:血浆cfdna样本的提取和测序方法:在ldct影像诊断前,对患者进行液态活检。采用紫色血液收集管(edta抗凝管)收集患者10ml全血样本,及时离心分离血浆(2小时内),在-80摄氏度冷冻保存下,转至实验室分析。转运至实验室后,血浆样本采用qiagen血浆dna提取试剂盒按照说明书进行ctdna提取。对采集到的cfdna样本建库后,进行wgs~5乘测序。在获得了下机数据之后,将数据比对至人类参考基因组上,获得相应的读段的碱基数据信息。
31.本专利的模型的建立过程主要如下:步骤2,对阳性组和对照组的样本进行cfdna的提取并测序,获得读据;
步骤3,将读段数据结果比对至参考基因组,获得在参考基因组上的不同窗口范围内的不同长度区间内的读段的数量,不同长度的读段数比值作为第一特征值;步骤4,将读段数据结果比对至参考基因组,得到读段的5’端在参考基因组上的位置;获得所述的位置处的上下游各m个bp碱基的序列数据,作为碱基片段集合;以得到的各个碱基片段在全部片段中的所占比例作为第二特征值;步骤5,将参考基因组划分为多个窗口,并分别获得在每个窗口范围内的拷贝数数据,作为第三特征值;步骤6,将读取到16bp 新短序列的读数和总读数的比例,作为第四特征;步骤7,分析所选取的转录因子的核小体覆盖模式,作为第五特征;步骤8,将阳性组和对照组的样本的模型特征向量输入第一层模型,选择出各个特征对应的5个auc最好的模型,第一层模型共选择出25个特征模型;步骤9,将步骤9选择出的25个模型输入至第二层整合模型,输出auc排序前三个整合模型,取这三个整合模型输出的预测可能性结果的平均值,作为最后判定结果;本专利中的特征值共五种,分别详述如下:1.dna片段大小比例(fragment size ratios, fsr)对于dna片段的大小比例,其反映的是cfdna读段的长度大小在良恶性肿瘤的分布特征。利用短dna片段和长dna片段的比例进行机器学习建立预测模型,后面(benign lungnodules)和恶性肺结节(malignant lungnodules);cfdna读段长度数据是通过如下方法获取得到的:在比对好的bam当中,记录了每一条读段的质量,长度和比对位置信息,人类参考基因组选用来自加利福尼亚大学克鲁兹分校(university of california, santa cruz, ucsc)提供的hg19序列。将人类参考基因组按照5mb长度,切割成541个窗口,分别统计每一个窗口中的短读段数量(100bp-151bp)和长读段数量(151bp-220bp),根据所有窗口中各种读段数量统计结果,分别对每种读段数量进行标准化换算,即标准化值=(原始值
–
平均值)/标准差。由此得到了541组不同长度的读段数量的数集。
32.2.读段5
‘
端断点处序列读段数量占比(breakpoint motifs,bpm) 人类参考基因组是dna双螺旋结构,依靠碱基互补配对氢离键链接;在正常衰老和癌症进展过程中,细胞周五环境的酸碱度发生变化,从而破坏了碱基互补氢键,发生断裂;由于断裂处的碱基序列不同,包含不同断点处序列的信息的序列占比也会不同。收集方法:比对后的bam中,记录了每一条读段的基本信息和比对到的位置,确认每一条读段的5
‘
端所在人类参考基因组序列坐标的断点左右各4bp序列,统计每种断点处的8bp长度序列(共计4**8=65536种)的读段数量,从而计算出65536种断点处序列读段占比,例aaaaaaaa读段占比 = aaaaaaaa读段数量 / 所有断点处序列读段总数。
33.3.1 mb窗口拷贝数变化(1 mb-bin copy number variation, cnv)拷贝数变化与个体癌症有显高度相关性,尽管已经可以通过检测部分癌症相关基因或特定基因组区间的拷贝数数变化从而进行区分,但仍有其他稀有或未知基因或区间可以提供潜在拷贝数变化信息。收集方法:首先收集30例健康人的wgs数据,将参考基因1-22号染色体以1mb的长度无重叠划分窗口,利用bedtools coverage对每个样本计算各个窗口内的读段深度,并根据各自窗口的gc含量和平均比对能力记录(ucsc bigwig文件)进行矫
正,取每个窗口中的30位健康人的中位深度作为代表,获得2475个窗口读段深度的群体对照基线;对每个待测样本,同样获得2475个窗口个体读段深度信息,利用隐马尔可夫模型(hidden markov model,hmm)和每个窗口群体对照基线深度,构建每个窗口的拷贝数变化对数,即log2(待测样本矫正均一化后深度/群体基线矫正均一化后深度),从而获得每个待测样本的拷贝数变化信息。
34.4.16bp 肿瘤新短序列 (16bp neomers, neo)无效子(nullomer)指人类基因组中不存在的dna短序列,16bp肿瘤新短序列是无效子的子集,特指在不出现在人类基因组里但在肿瘤组织基因组中反复发现的长度为16bp的无效体。
35.本专利中该特征值是通过如下方式进行获得:首先,使用穷举法生成所有可能的长度为16bp的短序列集合a。再在人类参考基因序列中(hg19版),以1bp为滑动窗口,使用穷举算法穷尽搜索所有长度为16bp的短序列集合b及其出现的次数,将仅集合a中出现的16bp短序列,定义为无效子。
36.本专利侧重于通过分析pcawg数据库(https://dcc.icgc.org/pcawg)中患有6种不同类型的种(肠癌,肺癌,乳腺癌,胃癌,前列腺癌,肝癌),获得这些不同类型的癌症的2577个患者的wgs突变结果,从中提取出977种多次出现的碱基替换(至少出现过两次)。根据碱基替换的位置,再使用穷举法提取出所有可能存在的包含这一碱基替换的无效子短序列的集合c。
37.通过gnomad(https://gnomad.broadinstitute.org/)数据库,收集东亚人群频率大于0.01的碱基替换的突变位点,根据这些突变位点的位置,再从无效子中找到包含这些位点的短序列集合d,作为收集东亚人群常见无效子集合。再从集合c中将出现过的集合d的无效子序列排除,最后得到4616个16bp肿瘤相关新短序列。针对这4616个新短序列,首先统计出样本中能够读取到任意一个4616个新短序列的样本数量,再针对每一个新短序列,搜索出包含这些新短序列的样本数量,将每一个新短序列的样本数与所有能读取到任意新短序列的样本总数的比例(共4616个比例数值),作为模型的第四特征。
38.5.核小体覆盖模式 (nucleosome coverage patterns, ncp)从gtrd数据库(https://gtrd.biouml.org/#!)(v21.12)中选择转录因子,排除掉不在cis-bp数据库(http://cisbp.ccbr.utoronto.ca/)(v2.00)中有已知转录位点的转录因子,共选择出334个有超过10000高匹配位点的转录因子。
39.对于以上获得的转录因子,以这些目标转录因子中转录位点附近-5kb到+5kb范围作为窗口,获得可以比对至这些窗口中的长度为100-220bp的片段。针对这些片段,进行gc矫正和对于测序深度使用多项式次方为3的savitzky-golay滤波器平缓曲线,得到最终每个转录因子的覆盖模式曲线。
40.在获得了上述的覆盖模式曲线后,针对每个转录因子,提取三个特征:1)对于转录因子的全部转录位点,求出这些转录位点的上端1kb到下端1kb的平均深度;2)转录因子的中心深度;3)对于上述获得 的覆盖模式曲线进行 快速傅里叶变换,获得核小体振幅信号的最高点的振幅数值。
41.将这三个特征共同作为核小体覆盖模式的特征值。
42.通过上述的数据获取,分别能够获得这五类数据的初始数据向量。接下来,再设计相应的计算方法,在本专利中可以采用常规的分类器算法通过上述的特征值进行分类,将这些特征值输入至分类器中,以发生恶性肺结节的概率值作为输出。也可以对已知的分类器进行优化调整,本专利中的优化所采用的分类器模型包括如下四种,并且在分类器优化时还同时生成了同一模型下的具有不同模型参数的子模型,进行子模型的筛选。四种主要的模型包括:1.弹性网络回归模型(elasticnetlogisticregression,glm)弹性网络回归模型是机器学习中常见的一类算法,是一个通过惩罚最大似然来拟合广义线性模拟的模型,他结合岭回归的l2正则化和lasso回归的l1正则化算法。正则化路径是针对正则化参数λ的值网格处的套索或弹性网络罚值计算的,解决了回归里的过拟合问题。超参数alpha控制正则化l1,l2的分布。
43.2.极致梯度提升(extremegradientboosting,xgboost)是一种基于梯度提升决策树(gradientboostingdecisiontree,gbdt)的集成思想加法模型的优化算法。他利用二阶泰勒公式展开,优化损失函数,提高计算精准度,利用正则项去简化模型,避免过拟合,采用blocks存储结构,可以并行计算。
44.本专利使用的xgboost中的及学习器为树模型。当树的深度增加,会增加树的复杂度,模型能得到更好的训练,也可能导致过拟合问题,超参数max_depth用于控制树的最大深度,超参数min_rows用于控制每个叶节点的最小样本数。
45.3.随机森林(randomforest)随机森林是一个强大的分类和回归工具,用于高维度和多重共线性的情况。当提供一组数据集合,随机森林可以随机抽取部分信息产生一组帮助分类或回归的决策树林,做节点分裂属性,不断重复随机抽取,直至不能再分裂;最后结合所有分裂属性结果,获得最终预测结果。随机森林也是通过超参数max_depth,min_rows等超参数来控制树的复杂度。
46.4.深度学习神经网络模型(deeplearning,nn)神经网络由输入,权重,偏差或阈值以及输出组成,任何单个节点的输出高于指定的阈值,则激活该节点,将数据发送到网络的下一层。输入层的每个节点,都要与隐藏层的每个节点做点对点的计算,运用加权求和和激活的方法。利用隐藏层计算出的每个值,再用相同的方法,和输出层进行计算。他具有分类准确度高,并行分布处理能力强,分布存储及学习能力强的优点。
47.本专利所使用的的深度学习神经网络模型为多层的前馈神经网络(multi-layerfeedforwardneuralnetwork),前馈网络的神经网络结构为最前面的输入层,中间的隐藏层,及最后的输出层,中间可含有多个,多层复杂的隐藏层(hiddenlayer),网络信号由输入层到输出层单向传输,每层的神经元仅与其前一层的神经元相连接受前一层的信息。深度学习神经网络采用随机梯度下降算法进行优化和训练,它使用训练数据集中的数据估计模型当前状态的误差梯度,然后利用误差更新模型的权重。在训练期间更新权重的量为学习率(epsilon),用于控制模型适应问题的速度,学习率衰退指数(rho),也用于控制模型学习的速率,都是神经网络训练中可配置的重要超参数。
48.另外,本专利还采用了随机搜索超参数 (random grid search parameters)算法,用于对模型进行优化。
49.随机搜索是一种常见的机器学习超参优化的方法。随机搜索就是在特定的模型参数范围之中随机抽取参数值,对多个抽样的参数值中选取最优的参数组合。该方法不是尝试所有可能的组合,而是通过选择每一个超参数的一个随机值的特定数量的随机组合。随机搜索相较于使用人工调优和网络搜素进行模型调参,可以用较少的搜索次数达到比较良好的效果,提供了一种更高效的解决方法(特别是参数数量多的情况下)。
50.本专利中四种算法在实施过程中,具体是采用了以下的
……
算法类型(或者说明一下是调用了哪种算法工具包中)本专利使用的四种算法的超参数如下表所示:在获得247例恶性肺结节和60例良性肺结节的患者的上述五类初始数据信息后,将片段大小比例(fsr)统计结果作为输入值(每个样本的输入向量中包括541个读段片段大小比例构成的特征值),分别通过四种分类模型对恶性肺结节样本与良性肺结节样本进行分类,在进行筛选的过程中,还分别对这四类模型通过随机搜索超参数进行参数和结构的变化,作为子模型进行数据的训练和模型的建议,再选出该特征的五个最优子模型,在筛选时是以模型的训练集的auc曲线作为分类效果的指标;同样地,通过收集良性肺结节患者和恶性肺结节患者的dna片段5’端断点序列读段数据占比信息后,以dna片段5’末端断点出序列占比(65536种),作为输入值,通过四种分类模型分别通过五种特征对恶性肺结节样本与良性肺结节样本进行分类,从中选出该特征的五个最优子模型(具体的模型优化及超参数调整过程同上)。同样地,拷贝数变化(2475种),新短序列(4616种)和核小体覆盖模式(1002种)也分别作为输入值,通过分别四类模型分类,并为每个特征选择出最优的五个子模型(具体的模型优化过程同上)。通过上述的计算过程,共得到5
×
5=25个模型计算结果。在每个计算中,可以获得每个特征向量对于分类结果的贡献值。
51.各个特征选择出的5个最优模型(共计25个模型)分别是如下表所示:
每个特征所选的最优模型的贡献值排前的特征变量以及贡献值如下:1.dna片段大小比例 (fsr) 深度学习神经网络模型(deep learning,nn):
2. 5’端断点处序列 (bpm) 读段弹性网络回归模型(glm):读段弹性网络回归模型(glm):3.拷贝数量变化 (cnv) 深度学习神经网络模型(deep learning,nn)
4.16bp 肿瘤新短序列(neo)xgboost模型:肿瘤新短序列(neo)xgboost模型:5.核小体覆盖模式(ncp)xgboost模型:
为进一步提高分类器预测性能,对25种训练模型结果进行二次集合训练(stacking)。stacking是一种集成学习技术,通过对25个底层弱分类器(1
st-level base model)的再次进行元学习(2
nd-level meta-learning),收集每个底层分类器的特点,找到最优整合方式,从而提高模型预测性能。最终本专利stacking使用的训练算法为广义线性模型(generalized linear model,glm),极致梯度提升xgboost模型和深度学习回归模型。
52.其中3个最优的整合模型及特征模型重要性(variables importance)为下表所示:
其中auc最高的的最优stacking模型为广义线性模型(glm),通过联结函数建立响应变量的数学期望值与线性组合的预测变量之间的关系,将25种训练模型转化为最终线性方程:allstacked=intercept+a*cnvmodel1+b*cnvmodel2+c*cnvmodel3+d*cnvmodel4+e*cnvmodel5+f*bpmmodel1+g*bpmmodel2+h*bpmmodel3+i*bpmmodel4+j*bpmmodel5+k*fsrmodel1+l*fsrmodel2+m*fsrmodel3+n*fsrmodel4+o*fsrmodel5+p*neomodel1+q*neomodel2+r*neomodel3+s*neomodel4+t*neomodel5+u*ncpmodel1+v*ncpmodel2+w*ncpmodel3+x*ncpmodel4+y*ncpmodel5其中,intercept、a-i都是线性方程参数。
53.具体系数如下:
每种特征在不同的训练算法下均有一定的预测效果,二次集合训练单一特征对该特征的预测效果均有提升。最终针对训练集的预测结果auc高达0.9474,针对验证集的预测结果auc达到0.931,敏感性为85%,特异性为98.7%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1