一种团簇元素之间结合倾向性的计算方法与流程
1.本发明属于第一性原理计算领域,具体涉及一种合金材料内形成溶质原子团簇时各元素偏聚结合倾向性大小的计算方法。
背景技术:
2.金属材料在经过热处理以及辐照过程后,部分材料内部会产生偏聚相或溶质原子偏析,偏析有形态、成分、结构等差异,而往往因偏析形成的团簇或者析出相会影响材料的使用性能,如核电站使用的反应堆压力容器材料(rpv钢)在服役时因高温高压且受到粒子辐照作用,微区原子伴随缺陷演化迁移,形成纳米级原子团簇的偏析,导致材料发生脆化。对形成的偏析、团簇结构进行深入理解,可进一步发展针对此类缺陷的恢复技术,如辐照团簇的恢复。可见厘清材料内元素偏聚时的结合行为,有助于揭示团簇偏聚的微观机理。
3.由于金属材料中不同元素之间形成的结构差异较大,传统方法计算元素偏析时相互之间的亲和力需要确定温度、完整结构等多种条件,其目的为计算得到特定元素在特定结构中具体的结合能数值,此类工作往往为了获取更精确的数值结果而设定杂化的计算参数,对于实际形成的由多种元素偏聚形成的团簇,无法直接构建完整的团簇结构模型进行计算,特别因辐照形成的团簇是伴随由级联过程中形成的离位峰演化而来,其过程时间极短,原子间形成结合的条件难以确定。所以现有的计算方法难以获得合金材料内形成溶质原子团簇时各元素偏聚结合倾向性大小。
技术实现要素:
4.有鉴于此,为了克服现有技术的缺陷,本发明的目的是提供一种合金材料内形成溶质原子团簇时各元素偏聚结合倾向性大小的计算方法。
5.为了达到上述目的,本发明采用以下的技术方案:
6.一种团簇元素之间结合倾向性的计算方法,包括如下步骤:
7.确定材料内的元素成分与晶体结构类型;
8.构建盒子空间、晶胞结构与孤立原子;
9.计算将x元素原子加入至纯y元素晶格中引起的第一自由能增量,计算将y元素原子加入至纯x元素晶格中引起的第二自由能增量,计算两次自由能增量的平均值;
10.根据自由能增量的平均值判断x元素和y元素之间相互结合的倾向。
11.根据本发明的一些优选实施方面,所述第一自由能增量的计算过程如下:将盒子空间中的孤立原子替换成x元素原子,将中心晶胞位置中的所有原子替换成y元素原子,计算此时的体系自由能e1;
12.将孤立原子替换成y元素原子,将中心晶胞位置的某一个原子替换成x元素原子,计算此时的体系自由能e2;
13.体系自由能e1和体系自由能e2之间的差值为第一自由能增量。
14.根据本发明的一些优选实施方面,所述体系自由能e1根据如下公式计算得到:
15.e1=e
x
+e
str
+e
x~str
≈e
x
+(∑e
atoms1
)+e016.式中,e
x
、e
str
、e
x~str
、e
atoms1
、e0分别为x元素原子在体系中的能量、中间晶胞结构的能量、x元素原子与中心晶胞之间的相互作用势能、中心晶胞中各原子单独在体系中的自由能、体系中所有原子之间的相互作用能。
17.中心晶胞指的是空间中构建的晶胞,体系指整个空间中的全部。
18.根据本发明的一些优选实施方面,所述体系自由能e2根据如下公式计算得到:
19.e2=ey+e
str
+e
y~str
≈ey+(∑e
atoms2
)+e
x,0
20.式中,ey、e
y~str
、e
atoms2
、e
x,0
分别为y元素原子在体系中的能量、y原子与中心晶胞之间的相互作用势能、替换后中心晶胞位置各原子单独在体系中的自由能、x原子引入后体系中所有原子之间的相互作用能。
21.根据本发明的一些优选实施方面,所述第一自由能增量表示为:
22.e
2-e1≈e
x,0-e0。
23.根据本发明的一些优选实施方面,所述第一自由能增量的计算过程如下:将盒子空间中的孤立原子替换成y元素原子,将中心晶胞位置中的所有原子替换成x元素原子,计算此时的体系自由能e
′1;
24.将孤立原子替换成x元素原子,将中心晶胞的某一个原子替换成y元素原子,计算此时的体系自由能e
′2;
25.根据体系自由能e
′1和体系自由能e
′2之间的差值为第二自由能增量。
26.根据本发明的一些优选实施方面,所述第二自由能增量表示为:
27.e
′
2-e
′1≈e
y,0-e0。
28.根据本发明的一些优选实施方面,所述自由能增量的平均值e
ave
表示为:
[0029][0030]
根据本发明的一些优选实施方面,计算得到的所述自由能增量的平均值越低,表明x元素与y元素在所构建的晶体结构环境中结合得越稳定,即相同条件下二者之间的原子结合倾向程度越高。
[0031]
根据本发明的一些优选实施方面,构建盒子空间、晶胞结构与孤立原子采用materials studio软件。
[0032]
由于采用了以上的技术方案,相较于现有技术,本发明的有益之处在于:本发明的团簇元素之间结合倾向性的计算方法,通过计算相应元素在不同元素组成的结构环境下的能量增值可以表征二者元素之间结合形成结构的稳定性,最终通过对比能量增值可快速获得多元素之间的结合倾向的相对大小,为快速判断团簇偏聚结构的成分提供理论依据。
附图说明
[0033]
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0034]
图1为本发明实施案例中ni原子加入至bcc型fe晶格原子替换示意图;其中,(a)为
替换前,(b)为替换后;
[0035]
图2本发明实施案例中各元素之间的相互作用能量。
具体实施方式
[0036]
为了使本技术领域的人员更好地理解本发明的技术方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分的实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
[0037]
本发明的目的在于提供一种计算并对比材料在特定行为后形成偏聚时元素之间结合倾向的计算方法,包括如下方法:
[0038]
步骤1)、确定材料内的元素成分与晶体结构类型。
[0039]
步骤2)、使用materials studio软件构建一个足够大的立方盒子空间,盒子空间边长≥计算晶胞晶格常数的8倍,在盒子空间的中间构建一个晶胞体系(晶胞体系越大,计算越精确,但计算量越大),并且在盒子空间中添加一个孤立原子,要求该孤立原子距离中心晶胞足够远,该孤立原子与中心晶胞最近的原子间距离≥计算晶胞晶格常数的8倍,且具有良好的对称性,以忽略该孤立原子与中心晶胞(空间中心位置处构建的晶胞)之间的相互作用势能和优化计算量。
[0040]
步骤3)、计算x元素和y元素之间的相互结合倾向
[0041]
3.1)计算x元素原子加入至纯y元素晶格后引起的第一自由能增量
[0042]
将盒子空间中的孤立原子替换成x元素原子,将中心晶胞中的所有原子替换成y元素原子,使用软件中的castep模块计算得到此种情况下的体系自由能e1:
[0043]
e1=e
x
+e
str
+e
x~str
≈e
x
+(∑e
atoms1
)+e0[0044]
式中,e
x
、e
str
、e
x~str
、e
atoms1
、e0分别为x原子在体系中的能量、中心晶胞结构的能量、x元素原子与中心晶胞的相互作用势能(当间距足够大时可忽略为0)、中心晶胞中各原子单独在体系中的自由能、体系所有原子之间的相互作用能。
[0045]
随后将孤立原子替换成y元素原子,将中心晶胞的某一个原子替换成x元素原子,所替换的原子位置尽可能在晶胞的几何中心,再次计算此时的体系自由能e2:
[0046]
e2=ey+e
str
+e
y~str
≈ey+(∑e
atoms2
)+e
x,0
[0047]
式中,ey、e
y~str
、e
atoms2
、e
x,0
分别为y元素原子在体系中的能量、y元素原子与中心晶胞的相互作用势能(当间距足够大时可忽略为0)、上述替换后中心晶胞中各原子单独在体系中的能量、x元素原子引入后体系中所有原子之间的相互作用能。
[0048]
根据替换前后整体系统各原子单独在体系中计算的自由能相等,则有:
[0049]ex
+∑e
atoms1
=ey+∑e
atoms2
[0050]
那么将x元素原子加入至纯y元素晶格后引起的第一自由能增量可表示为:
[0051]e2-e1≈e
x,0-e0[0052]
其实际意义为纯y元素组成的结构对x元素的吸引能力的相对大小。
[0053]
3.2)计算y元素原子加入至纯x元素晶格后引起的第二自由能增量
[0054]
将盒子空间中的孤立原子替换成y元素原子,将中心晶胞中的所有原子替换成x元
素原子,使用软件中的castep模块计算此种情况下的体系自由能e
′1。
[0055]
随后将孤立原子替换成x元素原子,将中心晶胞的某一个原子替换成y元素原子,计算得到此时的体系自由能e
′2。体系自由能e
′1和e
′2的计算与步骤3.1)类似,不再赘述。
[0056]
那么将y元素原子加入至纯x元素晶格后引起的第二自由能增量可表示为:
[0057]e′
2-e
′1≈e
y,0-e0[0058]
其实际意义为纯x元素组成的结构对y元素的吸引能力的相对大小。
[0059]
3.3)计算两次自由能增量的平均值e
ave
:
[0060][0061]
自由能增量的平均值为x元素与y元素结合倾向的数值表征,该值越低,表明x元素与y元素在所构建的晶体结构环境中结合得越稳定,即相同条件下二者原子之间的结合倾向程度越高。
[0062]
基于上述方法可以便捷地计算固体材料中形成元素偏聚或团簇时,原子之间的结合倾向,以便于更准确地剖析偏聚相或偏聚团簇的成分结构,为材料组织性能优化及创新提供理论指导。
[0063]
实施案例:
[0064]
本实施例对a508-3合金在辐照后形成的mn-ni-si型溶质原子团簇中的元素进行结合倾向性的计算,步骤如下:
[0065]
步骤1)、确定材料内形成溶质原子团簇的主要元素成分为fe、ni、mn、si,主要晶体结构为bcc型;
[0066]
步骤2)、在materials studio软件中构建边长为2nm的立方体盒子,在盒子中心位置构建2
×2×
2级bcc型晶胞,晶格常数为0.287nm,以及在盒子角落位置(0.2nm,0.2nm,0.2nm)处添加一个孤立原子a。
[0067]
步骤3)、计算ni原子加入至fe晶格后引起的自由能增量
[0068]
如图1所示,将a原子替换成ni,将bcc结构中的所有原子替换成fe,使用软件中的castep模块计算得到此种情况下的体系自由能e1=-9097.22ev。图1(a)为替换前的示意图;图1(b)为ni原子加入至bcc型fe晶格原子替换后的示意图。
[0069]
随后将a原子替换成fe,将bcc晶胞中心位置原子替换成ni,再次计算得到体系自由量e2=-9092.94ev。
[0070]
得到自由能增量为e
2-e1=4.28ev。
[0071]
步骤4)、计算fe原子加入至ni晶格后引起的自由能增量
[0072]
将a原子替换成fe,将bcc结构中的所有原子替换成ni,使用软件中的castep模块计算此种情况下的体系自由能e
′1=-13022.23ev。
[0073]
随后将a原子替换成ni,将bcc晶胞中心位置原子替换成fe,再次计算得到体系能量e
′2=-13027.56ev。
[0074]
得到自由能增量为e
′
2-e
′1=-5.33ev。
[0075]
步骤5)、计算两次自由能增量的平均值为e
ave
=-0.53ev。
[0076]
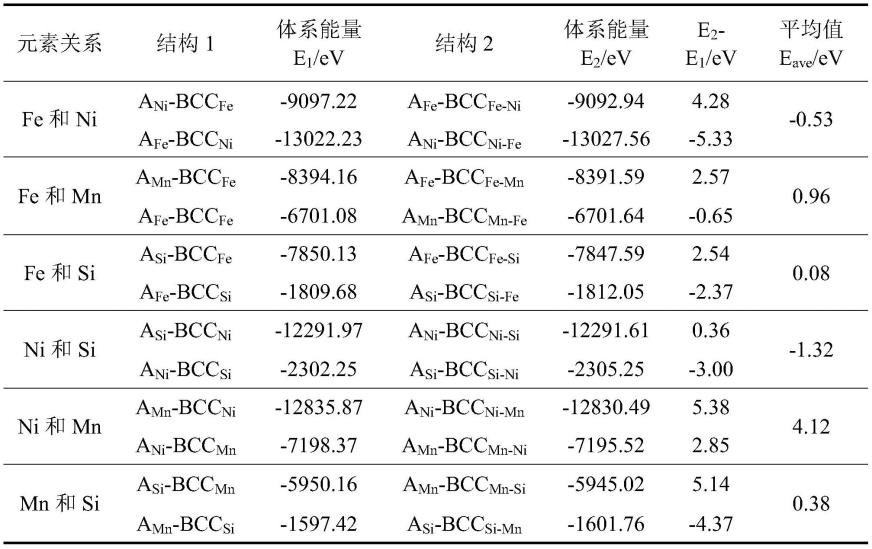
步骤6)、重复上述步骤,分别计算两两元素之间的自由能增量的平均值,见表1,并作图对比(如图2所示),得到团簇中上述元素之间的结合倾向为ni-si>fe-ni>fe-si>
si-mn>fe-mn>ni-mn。
[0077]
表1fe、ni、mn、si之间形成结构的能量关系
[0078][0079]
本发明的团簇元素之间结合倾向性的计算方法,通过计算相应元素在不同元素组成的结构环境下的能量增值可以表征二者元素之间结合形成结构的稳定性,最终通过对比能量增值可快速获得多元素之间的结合倾向的相对大小,为快速判断团簇偏聚结构的成分提供理论依据。
[0080]
上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1