一种基于RNA测序的癌症溯源方法
一种基于rna测序的癌症溯源方法
技术领域
1.本发明涉及肿瘤溯源技术领域,具体涉及一种基于rna测序的癌症溯源方法。
背景技术:
2.不明原发性癌症(cancerofunknownprimary,cup)是一种转移性肿瘤,是指尽管进行了标准的诊断检查,但仍然无法找到原发部位的一种疾病。如果不能准确了解患者的原发癌症部位,就不能对患者采取针对性的治疗方法,导致患者不能得到有效的治疗,让患者处于不利地位。所以,准确识别出癌症的原发部位对于患者的治疗是至关重要的。
3.目前,各种癌症溯源的方法所采用的数据种类有很多,比如基因表达数据、dna甲基化数据等等。其中,基于rna测序数据的方法主要有scope、cup-ai-dx、tod-cup等等。由于rna测序数据达到了上万维,这种具有高维特征的特性使得设计合适的机器学习方法变得困难。现有的多种机器学习方法存在模型简单或丢失原始基因信息的问题,比如scope使用简单的全连接网络来预测癌症类别,而简单的全连接网络可能无法有效地捕获基因之间的相互作用关系,这在一定程度上会影响分类性能。cup-ai-dx将卷积模块引入到了网络中,在网络设计上具有一定的创新,但是cup-ai-dx并不是使用了所有基因,而是提前进行了筛选,这种方法会导致一定程度上丢失原始的基因信息,影响分类性能。因此,为了进一步提升癌症分类的精确度,可以采用更强大的网络结构以有效捕获基因间的相互作用关系,同时使用所有的基因,避免基因筛选带来的丢失原始基因信息的问题。
技术实现要素:
4.本发明的目的在于提供一种基于rna测序的癌症溯源方法。
5.为实现上述目的,本发明采用以下技术方案:
6.一种基于rna测序的癌症溯源方法,包括以下步骤:
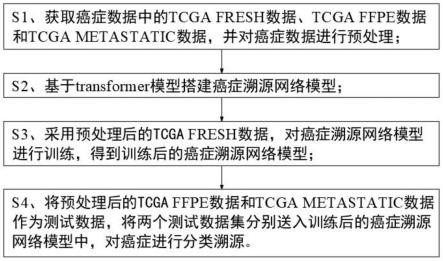
7.s1、获取癌症数据中的tcgafresh数据、tcgaffpe数据和tcga metastatic数据,并对癌症数据进行预处理;
8.s2、基于transformer模型搭建癌症溯源网络模型;
9.s3、采用预处理后的tcgafresh数据,对癌症溯源网络模型进行训练,得到训练后的癌症溯源网络模型;
10.s4、将预处理后的tcgaffpe数据和tcgametastatic数据作为测试数据,将两个测试数据集分别送入训练后的癌症溯源网络模型中,对癌症进行分类溯源。
11.优选地,步骤s1的具体过程为:
12.s11、从肿瘤基因组图谱tcga中,获取tcgafresh数据、tcgaffpe数据和tcgametastatic数据,癌症类型一共为32种;其中,tcgafresh数据和tcgaffpe数据采样来自原发灶,tcgametastatic数据采样来自转移灶,三种数据的格式都为fpkm;
13.s12、对于每个基因g,如果有一半以上的样本的基因g的表达值为0,则将该基因g删除;再对处理后的所有样本的基因数据进行log转换,具体操作为log(t+1),得到转换后
的数据,其中t为基因g的表达值;
14.s13、对每个癌型分配一个标签,一共有32种癌型,标签数从0到31,每个癌型对应一个数字标签;
15.s14、标签分配好后,根据每个样本所属的癌型为每个样本分配标签。
16.优选地,步骤s11中所述tcgafresh数据、tcgaffpe数据和tcga metastatic数据的样本数分别为9697、246和391。
17.优选地,步骤s2的具体过程为:
18.s21、使用一维卷积对rna测序数据进行处理,将原始rna数据x转换为适合transformer模型处理的数据xe,其中,x∈r
1xw
,xe∈r
nxp
,w为基因个数,p为每个嵌入表示的维数,为256维,n=w/p,每个卷积得到的结果为嵌入表示;
19.s22、将可学习的类别嵌入表示与卷积得到的嵌入表示进行拼接;
20.s23、将拼接后的嵌入表示与对应的位置嵌入表示进行相加,保留位置信息,得到输入数据;
21.s24、将输入数据输入到编辑器transformerencoder中,编辑器transformerencoder由多头自注意力模块和mlp模块组成,并在这两个模块前应用layernorm操作,且每个模块都用残差连接,编辑器transformerencoder的层数一共为4层,
22.s25、得到可学习的类别嵌入表示在编辑器transformerencoder对应的输出表示,将得到的结果输入到两层全连接网络中,获得32维输出,每维为各个癌症类别的概率,利用真实标签与transformer模型输出的概率计算交叉熵损失函数并优化transformer模型,得到癌症溯源网络模型;所述损失函数为:
[0023][0024]
其中,k为类别数,y为真实标签,如果类别为i,则yi为1,否则yi为0,y'表示网络输出的癌症的概率,yi'表示网络输出的第i个类癌症的概率。
[0025]
优选地,步骤s3的具体过程为:采用预处理后的tcgafresh数据作为训练数据,将其输入到癌症溯源网络模型中,再采用sgd算法作为模型优化算法,学习率设置为0.005,batchsize设置为128,训练迭代次数设置为100,当癌症溯源网络模型的损失函数值连续30个迭代没有下降时停止训练,并将最后得到的癌症溯源网络模型保存下来,得到训练后的癌症溯源网络模型。
[0026]
优选地,步骤s4还包括训练后的癌症溯源网络模型对tcgaffpe数据的嵌入表示进行t-sne可视化处理,将同一种癌症类别的样本聚集在一起,将不同种癌症类别的样本分开在不同的区域。
[0027]
采用上述技术方案后,本发明具有如下有益效果:本发明将transformer模型应用到了两万维rna测序数据上,这种先进的深度学习网络框架可以更有效地捕获基因间的相互作用关系,学习到各个癌症的潜在表示信息,且本发明可以使用所有的基因作为输入,不会存在基因筛选导致的丢失原始基因信息的问题,可有效提高癌症溯源分类的精确度。
附图说明
[0028]
图1为本发明的流程图;
[0029]
图2为本发明的癌症溯源网络模型的结构示意图;
[0030]
图3为本发明的编辑器transformerencoder的结构示意图;
[0031]
图4为本发明的t-sne可视化处理的结果图。
具体实施方式
[0032]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0033]
实施例
[0034]
如图1至图4所示,一种基于rna测序的癌症溯源方法,包括以下步骤:
[0035]
s1、获取癌症数据中的tcgafresh数据、tcgaffpe数据和tcga metastatic数据,并对癌症数据进行预处理;
[0036]
步骤s1的具体过程为:
[0037]
s11、从肿瘤基因组图谱tcga中,获取tcgafresh数据、tcgaffpe数据和tcgametastatic数据,癌症类型一共为32种;其中,tcgafresh数据和tcgaffpe数据采样来自原发灶,tcgametastatic数据采样来自转移灶,三种数据的格式都为fpkm;
[0038]
步骤s11中所述tcgafresh数据、tcgaffpe数据和tcga metastatic数据的样本数分别为9697、246和391;
[0039]
s12、对于每个基因g,如果有一半以上的样本的基因g的表达值为0,则将该基因g删除;再对处理后的所有样本的基因数据进行log转换,具体操作为log(t+1),得到转换后的数据,其中t为基因g的表达值;
[0040]
s13、对每个癌型分配一个标签,一共有32种癌型,标签数从0到31,每个癌型对应一个数字标签;
[0041]
s14、标签分配好后,根据每个样本所属的癌型为每个样本分配标签;
[0042]
s2、基于transformer模型搭建癌症溯源网络模型;
[0043]
步骤s2的具体过程为:
[0044]
s21、使用一维卷积对rna测序数据进行处理,将原始rna数据x转换为适合transformer模型处理的数据xe,其中,x∈r
1xw
,xe∈r
nxp
,w为基因个数,p为每个嵌入表示的维数,为256维,n=w/p,每个卷积得到的结果为嵌入表示;
[0045]
s22、将可学习的类别嵌入表示与卷积得到的嵌入表示进行拼接;
[0046]
s23、将拼接后的嵌入表示与对应的位置嵌入表示进行相加,保留位置信息,得到输入数据;
[0047]
s24、将输入数据输入到编辑器transformerencoder中,编辑器transformerencoder由多头自注意力模块和mlp模块组成,并在这两个模块前应用layernorm操作,且每个模块都用残差连接,编辑器transformerencoder的层数一共为4层,
[0048]
s25、得到可学习的类别嵌入表示在编辑器transformerencoder对应的输出表示,将得到的结果输入到两层全连接网络中,获得32维输出,每维为各个癌症类别的概率,利用
真实标签与transformer模型输出的概率计算交叉熵损失函数并优化transformer模型,得到癌症溯源网络模型;所述损失函数为:
[0049][0050]
其中,k为类别数,y为真实标签,如果类别为i,则yi为1,否则yi为0,y'表示网络输出的癌症的概率,yi'表示网络输出的第i个类癌症的概率;
[0051]
s3、采用预处理后的tcgafresh数据,对癌症溯源网络模型进行训练,得到训练后的癌症溯源网络模型;
[0052]
步骤s3的具体过程为采用预处理后的tcgafresh数据作为训练数据,将其输入到癌症溯源网络模型中,再采用sgd算法作为模型优化算法,学习率设置为0.005,batchsize设置为128,训练迭代次数设置为100,当癌症溯源网络模型的损失函数值连续30个迭代没有下降时停止训练,并将最后得到的癌症溯源网络模型保存下来,得到训练后的癌症溯源网络模型;
[0053]
s4、预处理后的tcgaffpe数据和tcgametastatic数据作为测试数据,将两个测试数据集分别送入训练后的癌症溯源网络模型中,对癌症进行分类溯源。本发明采用的评价指标为准确率,对于tcgaffpe数据,准确率的值达到了98.78%;对于tcgametastatic数据,准确率的值达到了93.61%。对于tcgaffpe数据,将癌症溯源网络模型输出的cls representation得到,并对其进行t-sne可视化处理,将同一种癌症类别的样本聚集在一起,将不同种癌症类别的样本分开在不同的区域,癌症溯源分类结果如图4所示。
[0054]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1