枸橼酸爱地那非胶囊剂及其制备方法和用途与流程
枸橼酸爱地那非胶囊剂及其制备方法和用途
发明领域
1.本发明属于药物制剂技术领域,尤其涉及一种枸橼酸爱地那非胶囊剂及其制备方法和用途。
2.发明背景
3.枸橼酸爱地那非是一种环磷酸鸟苷(cyclic guanosine monophosphate,cgmp)特异的5型磷酸二酯酶(phosphodiesterase 5,pde5)的选择性抑制剂,其本身不能直接舒张海绵体,但能明显促进性兴奋下引起的正常和性功能低下的阴茎勃起功能,其勃起功能增加不是促进一氧化氮(nitric oxide,no)释放,但no又能促进其作用。当性刺激引起局部no释放时,爱地那非抑制pde5可增加海绵体内cgmp水平,松弛平滑肌,使血液流入海绵体。但在没有性刺激时,推荐剂量的爱地那非不起作用。
4.枸橼酸爱地那非在药物空间结构、分子对接模型、靶点选择特异性、体内吸收代谢、用药剂量等方面做出了全面革新改进,在男性体内可以更加高效、快速的通过人体内细胞屏障,从而快速进入药物靶位,发挥药效。同时因为爱地那非在体内的转运通畅,因此不会产生体内药物残留,与国外同类型药物相比,具有吸收代谢更好,安全性更好的特点。从药效上来说,临床数据也表明,枸橼酸爱地那非片可大幅提高阴茎插入成功率(从45.08%增加到89.62%)和性交完成率(从10.76%增加到78.21%);与治疗前比较,勃起功能指数显著提高(平均提高10分以上)。
5.并且,根据专利cn 115054585 a记载,含有枸橼酸爱地那非的药物片剂还可用于治疗阿尔兹海默症,起到治疗认知功能下降、精神症状和行为障碍、日常生活自理能力的逐渐下降等中枢神经系统变性病的功能。
6.目前上市的枸橼酸爱地那非的剂型为片剂,规格为30mg(以爱地那非计),推荐剂量为60mg(以爱地那非计),其胶囊剂未上市。
7.胶囊剂具有以下优点:1)可掩盖药物的不良臭味,提高药物稳定性;2)药物以粉末或颗粒状态充填于胶囊内,可使药物在体内迅速起效;3)剂量准确,制备工艺简单,成本较低。
8.目前,并未查询到关于枸橼酸爱地那非的胶囊剂的相关专利,查询到其他pde5抑制剂主要有:西地那非和他达拉非等。根据现有查询到的相关专利可知,目前胶囊制剂存在的问题主要包括:
9.1、内容物多,服药者的服药顺应性低。
10.其中,专利cn 112206213a公开了一种枸橼酸西地那非组合物及其制备方法,该申请采用热熔挤出工艺,药物活性成分占比20%。根据其描述,若按西地那非普通片的成人正常服用剂量为50mg,则单片胶囊内容物的重量高达约250mg,胶囊体积大(是市售5
#
胶囊剂体积的2.5~3.1倍),会极大程度增加吞咽的难度系数,降低患者依从性。
11.2、制备工艺复杂,耗时长,生产效率低。
12.其中,专利cn 105106170a公开了一种治疗男性阳痿的药物他达拉非组合物胶囊,其所用的制剂工艺是粉碎原料药后过80目筛再进行湿法制粒的传统方法。原料药粉碎过筛
处理耗时较长,同时物料会有损失,湿法制粒后物料干燥时间也较长(长达3h),导致工艺效率低。
13.同时,由于枸橼酸爱地那非属于弱碱性药物,在中性介质中或弱碱性介质中溶出会降低,进而有可能对生物利用度产生影响。
14.基于现有技术存在的问题,急切需要提供一种枸橼酸爱地那非胶囊剂,所述胶囊剂能降低制剂产品的杂质含量,提高稳定性;同时提高枸橼酸爱地那非的溶出度;并且具有崩解速度快、制备工艺简单,稳定性和用药顺应性好,生物利用度高的优势。
技术实现要素:
15.针对现有技术存在的缺陷与不足,本发明提供了一种枸橼酸爱地那非胶囊剂,所述胶囊剂利用特定的辅料组合,配合特定的制备工艺,制备的胶囊剂
①
杂质含量低(低至0.005%,显著低于杂质限度标准≤0.1%)、稳定性高;
②
崩解速度快(30s内完全崩散)、溶出度高(可达100%,显著高于溶出度限度标准≥85%),溶出行为良好;
③
装量差异小(低至
±
1.8%),均匀度高;
④
生物利用度高,与爱力士-30mg
×
2片的相对生物利用度相比,能提高25%以上。
16.为了实现上述目的,本发明采用了如下技术方案:
17.一种枸橼酸爱地那非胶囊剂,所述胶囊剂包括药物活性成分,所述药物活性成分为枸橼酸爱地那非和/或其药学上可接受的盐。
18.所述胶囊剂由囊壁材料包裹囊芯材料构成。
19.所述囊芯材料包括药物活性成分,所述药物活性成分为枸橼酸爱地那非和/或其药学上可接受的盐。
20.所述囊芯材料还包括崩解剂、填充剂、润滑剂中的任意一种或几种。
21.优选地,所述囊芯材料还包括酸性辅料。
22.优选地,所述酸性辅料选自枸橼酸、富马酸、酒石酸中的任意一种或几种;更优选地,所述酸性辅料为酒石酸。
23.优选地,所述胶囊剂由囊壁材料包裹囊芯材料构成,所述囊芯材料包括药物活性成分、酸性辅料、崩解剂、填充剂和润滑剂;
24.所述药物活性成分为枸橼酸爱地那非和/或其药学上可接受的盐;
25.所述酸性辅料选自枸橼酸、富马酸、酒石酸中的任意一种或几种。
26.优选地,所述酸性辅料占囊芯材料的质量百分含量为0.3~10wt%,优选为0.5~5.0wt%;更优选地,所述酸性辅料占囊芯材料的质量百分含量为2.0wt%。
27.所述药物活性成分(以枸橼酸爱地那非计)占囊芯材料的的质量百分含量为10.0~80.0wt%,优选为55~75wt%,更优选为70wt%。
28.所述崩解剂占囊芯材料的质量百分含量为2.0~8.0wt%,优选为5.0wt%。
29.所述填充剂占囊芯材料的质量百分含量为15~50wt%,优选为20wt%。
30.所述润滑剂占囊芯材料的质量百分含量为0.5~2.0wt%,优选为1.0wt%。
31.优选地,所述胶囊剂中,按质量百分数计,囊芯材料包括如下组分:药物活性成分10~80wt%、酸性辅料0.3~10wt%、崩解剂2.0~8.0wt%、填充剂15~50wt%、润滑剂0.5~2.0wt%。
32.所述崩解剂选自羧甲基淀粉钠、交联羧甲基纤维素钠、低取代羟丙基纤维素、交联聚维酮中的任意一种或几种,优选为交联羧甲基纤维素钠。
33.所述填充剂选自乳糖、微晶纤维素、预胶化淀粉、磷酸氢钙、碳酸钙中的任意一种或几种,优选为微晶纤维素。
34.所述润滑剂为硬脂酸镁、胶态二氧化硅、硬脂酸钙、滑石粉中的任意一种或几种,优选为硬脂酸镁。
35.优选地,所述囊芯材料还包括粘合剂。
36.优选地,所述粘合剂占囊芯材料的质量百分含量1.0~6.0wt%。
37.所述粘合剂选自羟丙基纤维素、羟丙基甲基纤维素、羧甲基纤维素钠、聚维酮、淀粉浆中的任意一种或几种,优选为聚维酮。
38.优选地,所述胶囊剂用于治疗阿尔茨海默症。
39.优选地,所述胶囊剂用于治疗勃起功能障碍。
40.本发明还提供一种所述胶囊剂的制备方法,包括如下步骤:
41.1)将所有组分材料混合均匀,得到混合物;
42.2)将步骤1)的混合物制备成粉末、颗粒、微丸或微片;
43.3)灌装。
44.优选地,当含有酸性辅料时,所述步骤1)过程为:将酸性辅料与药物活性成分混合均匀。
45.优选地,当含有粘合剂时,所述步骤1)过程为:将酸性辅料和粘合剂以喷雾方式与药物活性成分混合均匀。
46.本发明还提供一种所述胶囊剂在制备用于治疗阿尔兹海默症药物中的用途。
47.本发明还提供一种所述胶囊剂在制备用于治疗勃起功能障碍药物中的用途。
48.优选地,所述胶囊剂在37℃,ph6.8磷酸盐缓冲液中15min内的溶出度≥85%本发明有益效果如下:
49.本发明提供了一种枸橼酸爱地那非胶囊剂,利用特定的产品处方,制备的胶囊剂具有多重优势:
50.①
杂质含量低(低至0.005%,显著低于杂质限度标准≤0.1%)、稳定性高;
51.②
崩解速度快(30s内完全崩散)、溶出度高(可达100%,显著高于溶出度限度标准≥85%),溶出行为良好;
52.③
装量差异小(低至
±
1.8%),均匀度高;
53.④
生物利用度高,与爱力士-30mg
×
2片的相对生物利用度为128.3%。
54.(一)酸性辅料
55.1)本发明在胶囊剂的囊芯材料中通过添加酸性辅料,制备得到的胶囊剂杂质含量显著降低、进而使稳定性显著提升;崩解时间短,溶出行为显著提高。
56.当选用的酸性辅料为酒石酸、富马酸或枸橼酸时,所得胶囊剂的稳定性和溶出行为良好,具体表现为:
57.i.稳定性良好:各杂质含量显著降低(0.005~0.05%,显著低于杂质限度标准≤0.1%);
58.ii.溶出行为良好:溶出度高达98~100%,显著高于限度标准≥85%,崩解时间短
(30s完全崩散);
59.其中,酸性辅料为酒石酸时,所得胶囊剂的稳定性和溶出行为最好,具体表现为:
60.i.稳定性最好:各杂质含量最低(低至0.005%,显著低于杂质限度标准≤0.1%);
61.ii.溶出行为最好:崩解时间最短(30s完全崩散),溶出度最高(高达100%,显著高于溶出度限度标准≥85%)。
62.2)通过改变酸性辅料的用量,能显著降低各杂质含量,进而提高胶囊剂的稳定性;并且提高溶出度,改善溶出行为:
63.当酸性辅料的质量分数为0.3~10%时,所得胶囊剂稳定性和溶出行为良好,具体表现为:
64.i.稳定性良好:各杂质含量显著降低(0.005~0.09%,显著低于杂质限度标准≤0.1%);
65.ii.溶出行为良好:崩解时间短(30s完全崩散)、溶出度高(90~100%,显著高于溶出度限度标准≥85%);
66.其中,酸性辅料的质量分数为2.0%时,所得稳定性和溶出行为最好,具体表现为:
67.i.稳定性最好:各杂质含量最低(低至0.005%,显著低于杂质限度标准≤0.1%);
68.ii.溶出行为最好:崩解时间最短(30s完全崩散),溶出度最高(高达100%,显著高于溶出度限度标准≥85%)。
69.(二)崩解剂
70.1)通过改变崩解剂的用量,能降低胶囊剂完全崩散所需时间,提高溶出度,改善溶出行为:
71.当崩解剂的用量占囊芯材料质量百分含量的2.0~8.0wt%时,所得溶出行为良好,具体表现为:
72.i.溶出行为良好:溶出度为90~100%,高于限度标准≥85%,崩解时间短(30s~1min完全崩散);
73.其中,当崩解剂的用量占囊芯材料质量百分含量的5.0wt%时,胶囊剂的溶出行为最好,具体表现为:
74.i.溶出行为最好:溶出度最高(高达100%,显著高于溶出度限度标准≥85%),胶囊剂在30s内完全崩散。
75.2)通过改变崩解剂的种类,能提高胶囊剂的溶出度,改善溶出行为:
76.采用本发明所述的羧甲基淀粉钠、交联羧甲基纤维素钠、低取代羟丙基纤维素或交联聚维酮作为崩解剂,所得胶囊剂溶出行为良好,具体表现为:
77.i.溶出行为良好:崩解时间短(30s完全崩散),溶出度高(96~100%,显著高于溶出度限度标准≥85%);
78.其中,崩解剂采用交联羧甲基纤维素钠时,胶囊剂的溶出行为最好,具体表现为:
79.i.溶出行为最好:溶出度最高(高达100%,显著高于溶出度限度标准≥85%),胶囊剂在30s内完全崩散。
80.(三)填充剂
81.1)通过改变填充剂的用量,提高胶囊剂的崩解效果,提高溶出度,改善溶出行为,并且胶囊剂的装量差异小,均匀度高;
82.控制填充剂占囊芯材料质量百分含量的15~50wt%时,所得胶囊剂溶出行为良好,并且装量差异小,均匀度高;具体表现为:
83.i.溶出行为良好:溶出度高(96~100%,显著高于溶出度限度标准≥85%),胶囊剂在30s内完全崩散;
84.ii.装量差异小:所述胶囊剂内容物的装量差异
±
1.8~4.2%,远小于限度标准
±
5%;
85.其中,填充剂占囊芯材料质量百分含量的20wt%时,胶囊剂溶出行为最好,并且装量差异最小,均匀度最高;具体表现为:
86.i.溶出行为最好:溶出度最高(高达100%,显著高于溶出度限度标准≥85%),胶囊剂在30s内完全崩散;
87.ii.装量差异最小:所述胶囊剂内容物的装量差异低至
±
1.8%,远小于限度标准
±
5%。
88.2)通过改变填充剂的种类,能使胶囊剂的装量差异减小,均匀度提高:
89.采用本发明所述的乳糖、微晶纤维素、预胶化淀粉、磷酸氢钙或碳酸钙作为填充剂时,所得胶囊剂内容物的装量差异小,均匀度高;具体表现为:
90.i.装量差异小:所述胶囊剂内容物的装量差异
±
1.8~3.6%,小于限度标准
±
5%;
91.其中,填充剂为微晶纤维素时,胶囊剂内容物的装量差异最小,均匀度最高;具体表现为:
92.i.装量差异最小:所述胶囊剂内容物的装量差异低至
±
1.8%,远小于限度标准
±
5%。
93.(四)润滑剂
94.1)通过改变润滑剂的用量,能提高崩解效果和溶出度,改善溶出行为;并且减小胶囊剂的装量差异,提高均匀度;
95.控制润滑剂占囊芯材料质量百分含量的0.5~2.0wt%时,所得胶囊剂溶出行为良好;并且装量差异小,均匀度高;具体表现为:
96.i.溶出行为良好:溶出度高(97~100%,显著高于溶出度限度标准≥85%),胶囊剂在30s~1min内完全崩散;
97.ii.装量差异小:所述胶囊剂内容物的装量差异
±
1.8~4.3%,小于限度标准
±
5%;
98.其中,润滑剂占囊芯材料质量百分含量的1.0wt%时,胶囊剂溶出行为最好,并且装量差异最小,均匀度最高;具体表现为:
99.i.溶出行为最好:溶出度最高(高达100%,显著高于溶出度限度标准≥85%),胶囊剂在30s内完全崩散;
100.ii.装量差异最小:所述胶囊剂内容物的装量差异低至
±
1.8%,远小于限度标准
±
5%;
101.2)通过改变润滑剂的种类,能降低完全崩散所需时间,提高溶出度,改善溶出行为;并且提高胶囊剂的装量差异,提高均匀度;
102.采用本发明所述的硬脂酸镁、胶态二氧化硅、硬脂酸钙或滑石粉作为润滑剂时,所
得胶囊剂溶出行为良好;装量差异小,均匀度高;具体表现为:
103.i.溶出行为良好:溶出度高(96~100%,显著高于溶出度限度标准≥85%),胶囊剂在30s内完全崩散;
104.ii.装量差异小:所述胶囊剂内容物的装量差异
±
1.8~2.8%,小于限度标准
±
5%;
105.其中,润滑剂为硬脂酸镁时,胶囊剂溶出行为最好;装量差异最小,均匀度最高;具体表现为:
106.i.溶出行为最好:溶出度最高(高达100%,显著高于溶出度限度标准≥85%),胶囊剂在30s内完全崩散;
107.ii.装量差异最小:所述胶囊剂内容物的装量差异低至
±
1.8%,远小于限度标准
±
5%。
108.(五)综合性能
109.1)采用本发明产品处方,对不同胶囊内容物形态(颗粒、粉末、微丸、微片)以及不同药物活性成分(10.0~80.0wt%)的胶囊剂产品均适用,所得胶囊剂稳定性和溶出行为良好,并且装量差异小,均匀度高;具体表现为:
110.i.稳定性良好:各杂质含量显著降低(0.005~0.08%,显著低于杂质限度标准≤0.1%);
111.ii.溶出行为良好:崩解时间短(30s完全崩散)、溶出度高(89~100%,显著高于溶出度限度标准≥85%);
112.iii.装量差异小:所述胶囊剂内容物的装量差异
±
1.8~3.8%,小于限度标准
±
5%。
113.2)采用本发明制备枸橼酸爱地那非胶囊剂的方法,操作更加简便,生产效率更高,减少了生产成本;同时,生物利用度高,与爱力士-30mg
×
2片的相对生物利用度相比,能提高25%以上(可达28.3%),显著提高了患者的用药顺应性。
具体实施方式
114.下面通过实施例来进一步说明本发明。应当理解为:本发明的实施例仅仅是用于说明本发明而给出,而不是对本发明的限制,在本发明技术方案的前提下对本发明的简单改进均属于本发明的保护范围。
115.实施例1:
116.本实施例提供了一种枸橼酸爱地那非胶囊剂,胶囊剂由囊壁材料包裹囊芯材料构成。按质量百分含量计,囊芯材料包括如下组分:
117.表1
118.成分作用质量/g质量百分含量/wt%枸橼酸爱地那非药物活性成分8470酒石酸酸性辅料2.42.0聚维酮粘合剂2.42.0微晶纤维素填充剂2420交联羧甲基纤维素钠崩解剂65.0
硬脂酸镁润滑剂1.21.0
119.上述枸橼酸爱地那非胶囊剂的制备工艺包括以下步骤:
120.a)配制粘合剂:
121.第i溶液:将酒石酸(酸性辅料)和处方中1/2用量聚维酮(粘合剂)溶于水中,配制成第i溶液,第i溶液中酒石酸的质量含量为5wt%;
122.第ii溶液:将处方中剩余1/2聚维酮(粘合剂)溶于水中,配制成溶液,第ii溶液中聚维酮的质量含量为10%。
123.b)原料药喷雾制粒:
124.将原料药加入流化床,采用流化床喷雾制粒,将步骤a)得到的第i溶液喷入,流化床设置进风温度60℃,喷液速度18rpm,风量40m3/h,105℃测定水分5min,水分控制在5%以内。
125.c)预混:
126.将微晶纤维素(填充剂)和交联羧甲基纤维素钠(崩解剂)与步骤b)喷雾制粒得到的颗粒混合均匀得到预混料;其中,混合转速为10rpm,混合时间为20min。
127.d)一步制粒:
128.将预混料加入流化床进行一步制粒,将步骤a)得到的第ii溶液喷入,设置进风温度60℃,喷液速度18rpm,风量40m3/h,105℃测定水分5min,水分控制在5%以内。
129.e)总混:
130.步骤d)制粒后颗粒用20目筛进行整粒,然后与硬脂酸镁(润滑剂)混合均匀,得到总混颗粒,其中,混合的转速为10rpm,混合的时间为10min。
131.f)灌装:
132.将总混颗粒使用5
#
胶囊模具进行填充、灌装,单个胶囊内容物的重量约为0.1g,单个即可达到推荐剂量为60mg(以爱地那非计)。
133.实施例1组分中添加了酸性辅料进行胶囊剂的制备;为了探究酸性辅料添加的必要性,实施例2省略酸性辅料以及制备工艺对应部分,然后进行一步制粒制备胶囊剂。
134.实施例2是否添加酸性辅料组分的影响
135.在实施例1的基础上,省略酸性辅料酒石酸以及制备过程中对应部分。处方中其他组分、处方用量和制备工艺同实施例1。
136.其制备工艺包括以下步骤:
137.a)配制粘合剂:
138.将聚维酮(粘合剂)溶于水中,配制成固含量为10%的溶液。
139.b)预混:
140.将药物活性成分、填充剂和崩解剂混合均匀得到预混料;其中,混合转速为10rpm,混合时间为20min。
141.c)一步制粒:
142.将预混料加入流化床进行一步制粒,设置进风温度60℃,喷液速度18rpm,风量40m3/h,105℃测定水分5min,水分控制在5%以内。
143.d)总混:
144.步骤c)制粒后颗粒用20目筛进行整粒,然后与润滑剂混合均匀,得到总混颗粒,其
中,混合的转速为10rpm,混合的时间为10min。
145.e)灌装:
146.将总混颗粒使用5
#
胶囊模具进行填充、灌装。
147.实施例3酸性辅料种类的选择
148.参照实施例1的处方,与实施例1的区别在于,将实施例1中作为酸性辅料的酒石酸,更换为:
149.方法1:富马酸;
150.方法2:枸橼酸;
151.方法3:乳酸;
152.方法4:醋酸;
153.处方中其他组分、处方用量和制备工艺同实施例1。
154.实施例4酸性辅料用量的选择
155.参照实施例1的处方,与实施例1的区别在于,将实施例1中酸性辅料酒石酸的用量2.0%,更换为:
156.方法1:0.3%;
157.方法2:10.0%;
158.方法3:0.1%;
159.方法4:15.0%;
160.处方中其他组分、处方用量和制备工艺同实施例1。
161.实施例5崩解剂用量的选择
162.参照实施例1的处方,与实施例1的区别在于,将实施例1中崩解剂交联羧甲基纤维素钠的用量为5%,更换为:
163.方法1:2%;
164.方法2:8%;
165.方法3:1.5%;
166.处方中其他组分、处方用量和制备工艺同实施例1。
167.实施例6崩解剂种类的选择
168.参照实施例1的处方,与实施例1的区别在于,将实施例1中作为崩解剂的交联羧甲基纤维素钠,更换为:
169.方法1:低取代羟丙基纤维素;
170.方法2:羧甲淀粉钠;
171.方法3:交联聚维酮;
172.方法4:交联羧甲基纤维素钠和交联聚维酮;
173.处方中其他组分、处方用量和制备工艺同实施例1。
174.实施例7填充剂用量的选择
175.参照实施例1的处方,与实施例1的区别在于,将实施例1中填充剂微晶纤维素的用量为20%,
176.更换为:
177.方法1:15%;
178.方法2:50%;
179.方法3:10%;
180.方法4:55%;
181.处方中其他组分、处方用量和制备工艺同实施例1。
182.实施例8填充剂种类的选择
183.参照实施例1的处方,与实施例1的区别在于,将实施例1中作为填充剂的微晶纤维素,
184.更换为:
185.方法1:乳糖;
186.方法2:预胶化淀粉;
187.方法3:磷酸氢钙;
188.方法4:碳酸钙;
189.处方中其他组分、处方用量和制备工艺同实施例1。
190.实施例9润滑剂用量的选择
191.参照实施例1的处方,与实施例1的区别在于,将实施例1中润滑剂硬脂酸镁的用量为1%,
192.更换为:
193.方法1:0.5%;
194.方法2:2.0%;
195.方法3:0.3%;
196.方法4:2.5%;
197.处方中其他组分、处方用量和制备工艺同实施例1。
198.实施例10润滑剂种类的选择
199.参照实施例1的处方,与实施例1的区别在于,将实施例1中作为润滑剂的硬脂酸镁,
200.更换为:
201.方法1:胶态二氧化硅;
202.方法2:硬脂酸钙;
203.方法3:滑石粉;
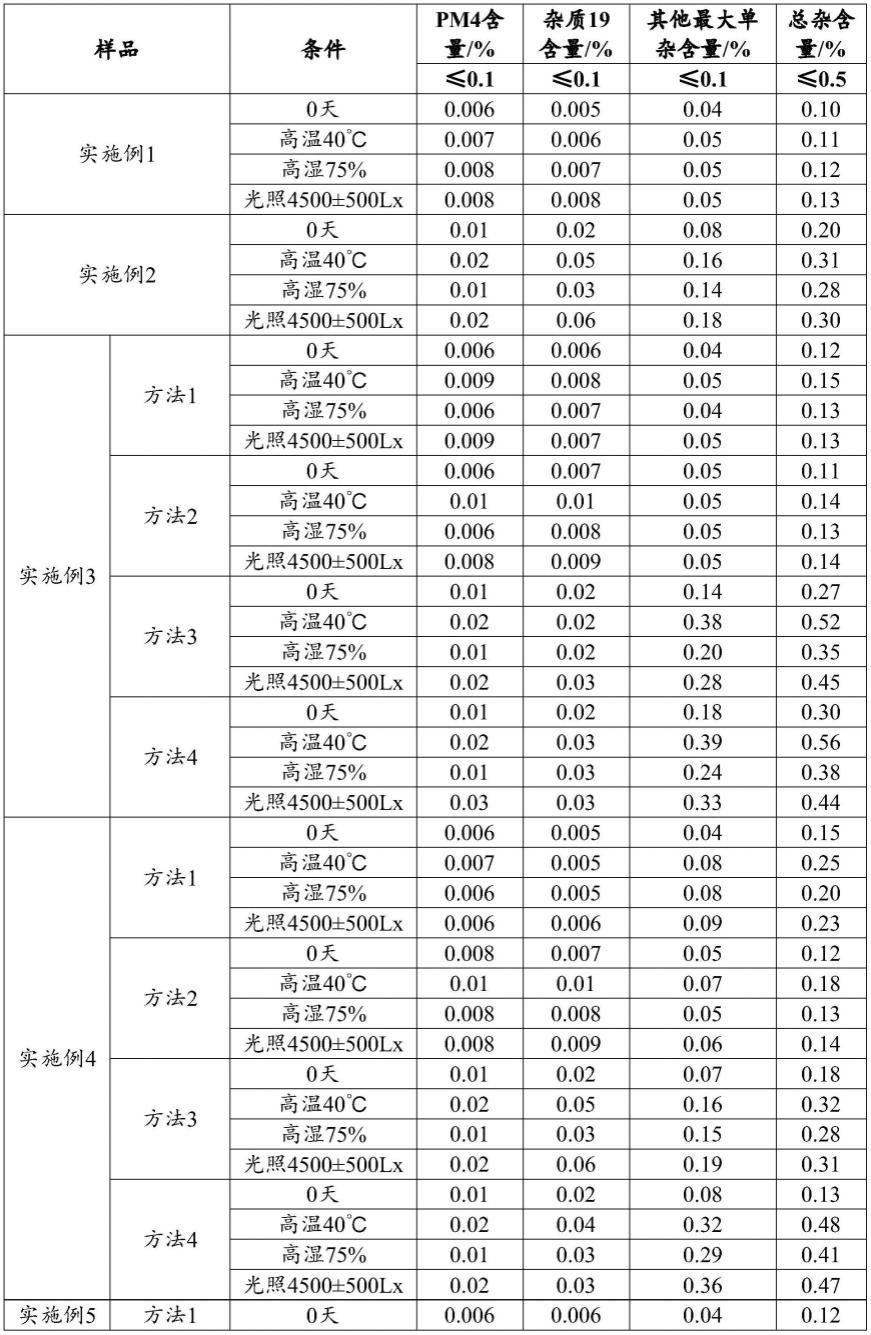
204.处方中其他组分、处方用量和制备工艺同实施例1。
205.为了探究胶囊剂内容物形态的影响,在实施例1胶囊剂的内容物(囊芯)为颗粒的基础上,通过改变制备方法获得不同形态的填装内容物,改变为粉末(实施例11方法1)以及不同方法制备得到的颗粒(实施例11方法2)、微丸(实施例11方法3和4)和微片(实施例11方法5)。
206.实施例11方法1采用原辅料直接混合粉末直接灌装工艺;
207.方法2先将原料药用酸性辅料和粘合剂的溶液喷雾制粒后,混合其他成分进行干法制粒然后灌装工艺;
208.方法3挤出滚圆上药制备微丸;
209.方法4空白丸芯上药制备微丸;
210.方法5挤压制备成微片。
211.实施例11不同胶囊内容物的制剂形态
212.方法1:胶囊内容物:粉末
213.参照实施例1的处方,采用将原辅料直接混合粉末直接灌装工艺,处方组分、处方用量同实施例1。
214.其制备工艺,包括以下步骤:
215.a)预混:
216.称取药物活性成分、酸性辅料、粘合剂、填充剂和崩解剂,将其混合均匀后,混合转速为10rpm,混合时间为20min,得预混料。
217.b)总混:
218.将预混料和润滑剂混合均匀后得总混颗粒,混合转速为10rpm,混合时间为10min;
219.c)灌装:
220.将总混颗粒使用5
#
胶囊模具进行填充、灌装。
221.方法2:胶囊内容物:颗粒
222.参照实施例1的处方,先将原料药用酸性辅料和粘合剂的溶液喷雾制粒后,混合其他成分进行干法制粒然后灌装工艺,处方组分、处方用量同实施例1。
223.a)配制粘合剂:
224.将酒石酸(酸性辅料)和聚维酮(粘合剂)溶于水中,配制成溶液,溶液中酒石酸的质量含量为5wt%。
225.b)原料药喷雾制粒:
226.将原料药加入流化床,采用流化床喷雾制粒,将粘合剂喷入,流化床设置进风温度60℃,喷液速度18rpm,风量40m3/h,105℃测定水分5min,水分控制在5%以内。
227.c)预混:
228.将微晶纤维素(填充剂)和交联羧甲基纤维素钠(崩解剂)与步骤b)喷雾制粒得到的颗粒混合均匀得到预混料;其中,混合转速为10rpm,混合时间为20min。
229.d)干法制粒:
230.利用干法制粒机进行干法制粒,筛网孔径为1.2mm,干法制粒参数如下:
231.表2
232.干法制粒优选参数送料螺旋转速/rpm60压辊转速/rpm10压辊间隙/mm1压辊压力/bar60整粒转速/rpm150
233.e)总混:
234.步骤d)制粒后颗粒用20目筛进行整粒,然后与硬脂酸镁(润滑剂)混合均匀,得到总混颗粒,其中,混合的转速为10rpm,混合的时间为10min。
235.f)灌装:
236.将总混颗粒使用5
#
胶囊模具进行填充、灌装。
237.方法3:胶囊内容物:微丸
238.参照实施例1的处方,将酸性辅料以粘合剂的形式与含枸橼酸爱地那非的微丸形式结合,然后灌装胶囊,其中微丸的上药方式为挤出滚圆上药,处方组分、处方用量除硬脂酸镁替换成微晶纤维素,其他同实施例1。
239.表3
240.成分作用质量/g质量百分含量/%枸橼酸爱地那非药物活性成分8470酒石酸酸性辅料2.42聚维酮粘合剂2.42微晶纤维素填充剂25.221交联羧甲基纤维素钠崩解剂65
241.a)配制粘合剂:
242.将酒石酸(酸性辅料)和聚维酮(粘合剂)溶于水中,配制成溶液,溶液中酒石酸的质量含量为5wt%。
243.b)预混:
244.将枸橼酸爱地那非、微晶纤维素和交联羧甲基纤维素钠混合均匀,混合20min,搅拌速度为10rpm,得预混料。
245.c)制备微丸:
246.在预混料中加入含酸性辅料的聚维酮溶液制备合适的软材,再在挤出滚圆机中制备微丸,干燥过筛即得微丸。
247.d)灌装:
248.将微丸使用5
#
胶囊模具进行填充、灌装。
249.方法4:胶囊内容物:微丸
250.参照实施例1的处方,将原料药、酸性辅料以粘合剂的形式与含微晶纤维素和交联羧甲基纤维素钠的空白丸芯相结合,然后灌装胶囊,其中微丸的上药方式为空白丸芯上药,处方组分、处方用量除硬脂酸镁替换成羟丙甲纤维素,其他同实施例1。
251.表4
252.成分作用质量/g质量百分含量/%枸橼酸爱地那非药物活性成分8460酒石酸酸性辅料2.82聚维酮粘合剂2.82微晶纤维素填充剂2820交联羧甲基纤维素钠崩解剂5.64羟丙甲纤维素粘合剂16.812
253.a)配制粘合剂:
254.将聚维酮(粘合剂)溶于水中,配制成溶液,溶液中聚维酮的质量含量为10%。
255.b)预混:
256.将微晶纤维素和交联羧甲基纤维素钠混合均匀,混合20min,搅拌速度为10rpm,得预混料。
257.c)制备空白丸芯:
258.在预混料中加入含粘合剂制备合适的软材,再在挤出滚圆机中制备空白丸芯,干燥过筛即得空白丸芯。
259.d)配制上药层溶液:
260.将枸橼酸爱地那非、酸性辅料加入适量水搅拌溶解后,再加入羟丙甲纤维素搅拌溶解配制成上药层溶液。
261.e)制备微丸:
262.将空白丸芯加入流化床内,底喷加入上药层溶液进行包衣,流化床设置进风温度60℃,喷液速度18rpm,风量40m3/h,105℃测定水分5min,水分控制在5%以内,出料过筛即得微丸。
263.f)灌装:
264.将微丸使用5
#
胶囊模具进行填充、灌装。
265.方法5:胶囊内容物:微片
266.参照实施例1的处方,在实施例11方法3的基础上,将步骤c)制备得到的微丸压成微片形态,然后经灌装,得到胶囊剂内容物为微片的胶囊剂产品。
267.实施例12不同药物活性成分的含量
268.参照实施例1的处方,与实施例1的区别在于,保证处方中药物活性成分总重量84g不变,改变填充剂的用量,将实施例1处方中药物活性成分的含量70.0wt%,调整为:
269.方法1:处方中药物活性成分的质量百分含量为10.0wt%;
270.方法2:处方中药物活性成分的质量百分含量为40.0wt%;
271.方法3:处方中药物活性成分的质量百分含量为80.0wt%;
272.活性成分比例的百分含量改变通过处方中的填充剂调整,其他组分、用量和制备工艺同实施例1。
273.实施例13枸橼酸爱地那非胶囊剂的应用
274.专利cn 115054585 a记载,含有枸橼酸爱地那非的药物片剂可用于治疗阿尔兹海默症。因此,将本发明制备的枸橼酸爱地那非胶囊剂用于治疗阿尔兹海默症,具有良好的治疗效果。
275.为验证本发明的效果,取上述实施例所制得的枸橼酸爱地那非胶囊剂进行质量检测对比。
276.实验例1:稳定性考察
277.按照高效液相色谱法(《中国药典》2020年版通则0512)测定以下实施例制备得到的制剂产品在新鲜制备条件(0天)以及分别在不同条件下放置30天的各杂质含量和稳定性情况,结果见下表。
278.其中,杂质pm4:工艺及降解杂质也是中间体pm4,化学名:4-(5-(((3s,5r)-3,5-二甲基哌嗪-1-基)磺酰基)-2-乙氧基苯甲酰氨基)-1-甲基-3-丙基-1h-吡唑-5-甲酰胺,结构式为:
[0279][0280]
杂质19:工艺及降解杂质,化学名:4-乙氧基-3-(1-甲基-7-氧代-3-丙基-6,7-二氢-1h-吡唑[4,3-d]嘧啶-5-基)苯磺酸,结构式为:
[0281][0282]
放置条件:
[0283]
1)高温40℃放置30天;
[0284]
2)高湿度条件为75%放置30天;
[0285]
3)光照强度为4500
±
500lx放置30天。
[0286]
表5
[0287]
[0288]
[0289]
[0290][0291]
实验例2:溶出度情况
[0292]
按照溶出度与释放度测定法(《中国药典》2020年版通则0931第二法)测定,同时观察溶出过程中溶出杯中样品状态,溶出介质为ph6.8磷酸盐缓冲液,结果如下。
[0293]
表6
[0294]
[0295][0296]
实验例3:胶囊剂内容物溶解过程中微环境ph
[0297]
胶囊剂的产品处方中添加酸性辅料能显著提高溶出及稳定性,因此为验证添加酸性辅料改善稳定性和溶出行为的相关机理,分别检测实施例胶囊内容物中间体微环境中的ph值,结果发现中间体微环境的ph有显著性差异。
[0298]
研究发现,影响胶囊剂内容物溶解过程中微环境ph的因素主要是:酸性辅料添加
及其种类及用量的(实施例1~4);而其他辅料成分对胶囊剂内容物溶解过程中微环境ph(即,中间体ph)影响很小,因此对实施例1~4的中间体微环境的ph值进行进一步检测。
[0299]
试验过程为:分别将实施例中的胶囊剂内容物完全转移至含有10ml纯化水的试管中,超声溶解,检测中间体微环境中的ph,结果如下:
[0300]
表7
[0301][0302]
实验例4:胶囊的填充装量情况
[0303]
分别取上述实施例制备得到的胶囊产品20粒,进行装量差异检测(即,胶囊内容物之间的重量差异),颗粒均一性越好,颗粒流动性越好,填装越均匀,装量差异越小,即,胶囊之间的重量越均一,胶囊剂中药物活性成分含量越准确。反之,装量差异越大,即,胶囊之间的重量差异越高,胶囊剂中药物活性成分含量差异越大。装量差异的限度标准为
±
5%。
[0304]
表8
[0305]
[0306][0307]
实施例7方法3:填充剂微晶纤维素用量仅为10%,用量<15%,微晶纤维素用量少,导致颗粒均一性较差,装量差异超出限度标注(
±
5%)。
[0308]
实施例9方法3:润滑剂硬脂酸镁用量仅为0.3%,用量<0.5%,硬脂酸镁用量少,导致物料流动性较差,灌装不顺畅,装量差异超出限度标注(
±
5%)。
[0309]
结果分析:
[0310]
对上述实施例的胶囊稳定性、溶出度结果、内容物溶解过程中微环境ph以及填充装量情况进行分析,探究胶囊剂组分中酸性辅料、填充剂、崩解剂和润滑剂的种类及其用量,以及制备工艺和胶囊内容物的形态与对胶囊剂的影响。
[0311]
(一)、为了探究胶囊剂组分中酸性辅料添加的必要性,将实施例1组分中添加了酸性辅料进行胶囊剂的制备,实施例2省略酸性辅料以及制备工艺对应部分,然后进行一步制粒制备胶囊剂,二者制备的胶囊剂进行比较。
[0312]
1、区别
[0313]
表9
[0314]
实施例酸性辅料实施例1酒石酸实施例2无
[0315]
2、制剂对比
[0316]
表10
[0317][0318]
表11
[0319]
样品溶出现象溶出度限度/≥85%实施例130s完全崩散,杯底无堆积100%实施例25min完全崩散,杯底少量堆积73%
[0320]
表12
[0321]
样品中间体ph实施例13.04实施例25.64
[0322]
表13
[0323]
样品装量差异/
±
%限度
±
5%实施例11.8实施例24.2
[0324]
结论:
[0325]
1)杂质含量结果显示:
[0326]
新鲜制备条件下(0天):
[0327]
实施例1(添加酸性辅料)制备得到的胶囊剂中杂质pm4含量为0.006%,杂质19含量为0.005%,其他最大单杂含量为0.04%,总杂含量为0.10%,均远低于杂质限度(pm4含量≤0.1%,杂质19含量≤0.1%,其他最大单杂含量≤0.1%,总杂含量≤0.5%)。
[0328]
实施例2(省略酸性辅料)制备得到的胶囊剂中杂质pm4含量为0.01%,杂质19含量为0.02%,其他最大单杂含量为0.08%,总杂含量为0.20%,虽然都控制在杂质限度标准(pm4含量≤0.1%,杂质19含量≤0.1%,其他最大单杂含量≤0.1%,总杂含量≤0.5%)之内,但是较实施例1(添加酸性辅料)各杂质含量均呈现增加趋势。
[0329]
高温40℃条件下放置30天:
[0330]
实施例1(添加酸性辅料)制备得到的胶囊剂中pm4含量为0.007%,杂质19含量为0.006%,其他最大单杂含量为0.05%,总杂含量为0.11%。较新鲜制备条件下(0天)的胶囊剂,各杂质增加量可忽略不计,并且均远低于杂质限度(pm4含量≤0.1%,杂质19含量≤0.1%,其他最大单杂含量≤0.1%,总杂含量≤0.5%),证明胶囊剂的稳定性良好且显著。
[0331]
实施例2(省略酸性辅料)制备得到的胶囊剂中杂质pm4含量为0.02%,杂质19含量为0.05%,总杂含量分别为0.31%;其他最大单杂含量为0.16%,超出限度标准(其他最大单杂含量≤0.1%),无法得到合格的胶囊剂产品。并且,较新鲜制备条件下(0天)的胶囊剂相比,实施例2(省略酸性辅料)高温40℃条件下放置30天杂质含量呈现大幅增加。其中,实施例2(省略酸性辅料)高温40℃条件下放置30天的杂质19含量是实施例1(添加酸性辅料)相同条件下的约8倍;各杂质含量均高于实施例1(添加酸性辅料),稳定性差于实施例1(添加酸性辅料)。
[0332]
高湿75%条件下放置30天:
[0333]
实施例1(添加酸性辅料)制备得到的胶囊剂中pm4含量为0.008%,杂质19含量为0.007%,其他最大单杂含量为0.05%,总杂含量为0.12%。较新鲜制备条件下(0天)的胶囊剂,各杂质增加均不明显,并且均远低于杂质限度(pm4含量≤0.1%,杂质19含量≤0.1%,其他最大单杂含量≤0.1%,总杂含量≤0.5%),证明胶囊剂的稳定性良好且显著。
[0334]
实施例2(省略酸性辅料)制备得到的胶囊剂中,pm4含量为0.01%,杂质19含量为0.03%,总杂含量为0.28%;其他最大单杂含量为0.14%,超出限度标准(其他最大单杂含量≤0.1%),无法得到合格的胶囊剂产品。
[0335]
光照4500
±
500lx条件下放置30天:
[0336]
实施例1(添加酸性辅料)制备得到的胶囊剂中pm4含量为0.008%,杂质19含量为0.008%,其他最大单杂含量为0.05%,总杂含量为0.13%。较新鲜制备条件下(0天)的胶囊剂,各杂质含量均无显著增加,并且仍保持远低于杂质限度标准(pm4含量≤0.1%,杂质19含量≤0.1%,其他最大单杂含量≤0.1%,总杂含量≤0.5%),证明胶囊剂的稳定性良好且
显著。
[0337]
实施例2(省略酸性辅料)制备得到的胶囊剂中pm4含量为0.02%,杂质19含量为0.06%,总杂含量为0.30%;其他最大单杂含量为0.18%,超出限度标准(其他最大单杂含量≤0.1%),无法得到合格的胶囊剂产品。
[0338]
上述结果表明,实施例1(添加酸性辅料)比实施例2(省略酸性辅料)制备得到的胶囊剂,稳定性更高。
[0339]
2)溶出行为结果显示:
[0340]
实施例1(添加酸性辅料)得到的制剂在30s完全崩散,杯底无堆积,溶出度为100%,远高于溶出度限度(≥85%),溶出行为良好。
[0341]
实施例2(省略酸性辅料)的制剂在5min内完全崩散,崩解时间显著增加(增加为实施例1的10倍),杯底出现堆积,溶出度为73%,不符合溶出度限度标准(≥85%)。溶出行为差。
[0342]
上述结果表明,只有实施例1(添加酸性辅料)制备的胶囊剂溶出效果显著。
[0343]
3)胶囊剂内容物溶解过程中微环境ph结果显示:
[0344]
实施例1组分中添加酸性辅料制备得到的胶囊内容物,在溶解过程中微环境ph为3.04。实施例2组分中省略酸性辅料制备得到的胶囊内容物,在溶解过程中微环境ph为5.64。实施例1组分中添加酸性辅料后,微环境ph下降,使得药物活性成分更易溶,进而显著提高胶囊剂的溶出度,进一步佐证了上述溶出行为结果的准确性。
[0345]
4)胶囊填装结果显示:
[0346]
实施例1组分中添加酸性辅料制备得到的胶囊内容物,颗粒流动性好,填充的重量更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0347]
实施例2组分中省略酸性辅料制备得到的胶囊内容物,胶囊内容物的装量差异为
±
4.2%,呈现增加趋势,但控制在限度标准
±
5%范围内。
[0348]
上述结果表明,实施例1添加酸性辅料制备得到的胶囊剂,能得到内容物的装量差异显著优于限度标准的胶囊剂。
[0349]
小结:
[0350]
①
采用本发明酸性辅料(例如,枸橼酸、富马酸、酒石酸)制备得到的胶囊剂,能制备稳定性和溶出行为良好胶囊剂;
[0351]
例如,实施例1采用酒石酸制备得到的胶囊剂,制备的胶囊剂稳定性溶出行为良好,具体表现为:
[0352]
所述胶囊中各杂质含量低至0.005%,显著低于杂质限度标准≤0.1%,稳定性良好;并且胶囊剂的溶出度高达100%,显著高于限度标准≥85%,崩解速度快(30s完全崩散),溶出行为显著。
[0353]
②
而省略酸性填料(例如,实施例2)得到的胶囊剂稳定性和溶出行为差,无法得到合格的胶囊剂,具体表现为:
[0354]
所述胶囊剂放置后的其他最大单杂含量高达0.18%,超出限度标准(其他最大单杂含量≤0.1%);并且胶囊剂完全崩散时间延长至5min,并且溶出度仅为73%,远低于限度标准≥85%,无法制备合格的胶囊剂。
[0355]
(二)以上结果分析(一)和(二)部分表明,在胶囊剂中添加酸性辅料制备的胶囊剂
效果最好(实施例1),因此,以下结果分析在实施例1的基础上,改变酸性辅料的种类,探究酸性辅料的种类对胶囊剂质量的影响:
[0356]
1、区别
[0357]
表14
[0358]
实施例酸性辅料实施例1酒石酸实施例3方法1富马酸实施例3方法2枸橼酸实施例3方法3乳酸实施例3方法4醋酸
[0359]
2、制剂对比
[0360]
表15
[0361][0362]
表16
[0363][0364]
表17
[0365][0366]
表18
[0367][0368]
结论:
[0369]
1)杂质含量结果显示:
[0370]
新鲜制备条件下(0天):
[0371]
实施例1(酸性辅料=酒石酸)、实施例3方法1(酸性辅料=富马酸)和实施例3方法2(酸性辅料=枸橼酸)的pm4含量均为0.006%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.007%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.10~0.12%,远低于限度标准(≤0.5%),稳定性良好。
[0372]
实施例3方法3(酸性辅料=乳酸)的pm4含量为0.01%;杂质19含量为0.02%;总杂含量为0.27%;各杂质含量均高于实施例1(酸性辅料=酒石酸)的胶囊剂产品。其他最大单杂含量显著增加达0.14%,不符合限度标准(≤0.1%),稳定性差,无法制备合格的胶囊剂产品。
[0373]
实施例3方法4(酸性辅料=醋酸)的pm4含量为0.01%;杂质19含量为0.02%;总杂含量为0.30%;各杂质含量均高于实施例1(酸性辅料=酒石酸)的胶囊剂产品。其他最大单杂含量显著增加达0.18%,不符合限度标准(≤0.1%),稳定性差,无法制备合格的胶囊剂产品。
[0374]
高温40℃条件下放置30天:
[0375]
实施例1(酸性辅料=酒石酸)、实施例3方法1(酸性辅料=富马酸)和实施例3方法2(酸性辅料=枸橼酸)的pm4含量为0.007~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.01%,远低于限度标准(≤0.1%);其他最大单杂含量均为0.05%,远低于限度标准(≤0.1%);总杂含量为0.11~0.15%,远低于限度标准(≤0.5%),稳定性良好。
[0376]
实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸)的pm4含量均为0.02%;杂质19含量为0.02~0.03%;各杂质含量均高于实施例1(酸性辅料=酒石酸)的胶囊剂产品。并且其他最大单杂含量显著增加达0.38~0.39%,不符合限度标准(≤0.1%);总杂含量显著增加至0.52~0.56%,不符合限度标准(≤0.5%);稳定性差,无法制备合格的胶囊剂产品。
[0377]
高湿75%条件下放置30天:
[0378]
实施例1(酸性辅料=酒石酸)、实施例3方法1(酸性辅料=富马酸)和实施例3方法2(酸性辅料=枸橼酸)的pm4含量为0.006~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.007~0.008%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.12~0.13%,远低于限度标准(≤0.5%),稳定性良好。
[0379]
实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸)的pm4含量均为0.01%;杂质19含量分别为0.02%和0.03%;总杂含量显著增加至0.35%和0.38%;各杂质含量均高于实施例1(酸性辅料=酒石酸)的胶囊剂产品。并且其他最大单杂含量显著增加达0.20%和0.24%,不符合限度标准(≤0.1%);稳定性差,无法制备合格的胶囊剂产品。
[0380]
光照4500
±
500lx条件下放置30天:
[0381]
实施例1(酸性辅料=酒石酸)、实施例3方法1(酸性辅料=富马酸)和实施例3方法2(酸性辅料=枸橼酸)的pm4含量为0.008~0.009%,远低于限度标准(≤0.1%);杂质19含量为0.007~0.009%,远低于限度标准(≤0.1%);其他最大单杂含量均为0.05%,远低于限度标准(≤0.1%);总杂含量为0.13~0.14%,远低于限度标准(≤0.5%),稳定性良好。
[0382]
实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸)的pm4含量分别为0.02%和0.03%;杂质19含量均为0.03%;总杂含量显著增加至0.45%和0.44%;各杂质含量均高于实施例1(酸性辅料=酒石酸)的胶囊剂产品。并且其他最大单杂含量显著增加达0.28%和0.33%,不符合限度标准(≤0.1%);稳定性差,无法制备合格的胶囊剂产品。
[0383]
上述结果表明,实施例1(酸性辅料=酒石酸)、实施例3方法1(酸性辅料=富马酸)和实施例3方法2(酸性辅料=枸橼酸)制备得到的胶囊剂,其稳定性显著优于实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸);并且实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸)无法制备稳定性合格的胶囊剂产品。
[0384]
2)溶出行为结果显示:
[0385]
实施例1(酸性辅料=酒石酸)得到的制剂在30s完全崩散,杯底无堆积,溶出度为100%,远高于溶出度限度(≥85%),溶出行为良好且显著。
[0386]
实施例3方法1(酸性辅料=富马酸)和实施例3方法2(酸性辅料=枸橼酸)得到的制剂在30s完全崩散,杯底无堆积,溶出度均为98%,远高于溶出度限度(≥85%),溶出行为良好。
[0387]
实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸)得到的制剂虽然在30s完全崩散,但是杯底出现堆积,溶出度降低至为81%和83%,不符合溶出度限度标准(≥85%),溶出行为差,无法制备溶出行为合格的胶囊剂产品。
[0388]
上述结果表明,实施例1(酸性辅料=酒石酸)、实施例3方法1(酸性辅料=富马酸)和实施例3方法2(酸性辅料=枸橼酸)制备得到的胶囊剂,其溶出行为显著优于实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸);并且实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸)无法制备溶出行为合格的胶囊剂产品。
[0389]
3)胶囊剂内容物溶解过程中微环境ph结果显示:
[0390]
实施例1(酸性辅料=酒石酸)的胶囊内容物,在溶解过程中微环境ph为3.04。实施例3方法1(酸性辅料=富马酸)和实施例3方法2(酸性辅料=枸橼酸)微环境ph分别为3.16和3.46;微环境ph呈酸性条件,使得药物活性成分更易溶,进而显著提高胶囊剂的溶出度,并保持良好的稳定性。
[0391]
但是,实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸)的微环境ph分别为4.84和5.62;ph较实施例1(酸性辅料=酒石酸)显著升高,无法使药物活性成分充分的溶出,从而使溶出度下降。同时,由于乳酸和醋酸二者的稳定性差于本技术的酸性辅料种类(酒石酸、富马酸、枸橼酸),因此自身发生降解,导致有关物质的杂质含量增加,降低了胶囊剂的稳定性。进一步验证了上述部分1)杂质含量结果和2)溶出行为结果的准确度。
[0392]
4)胶囊填装结果显示:
[0393]
实施例1(酸性辅料=酒石酸)制备得到的胶囊内容物,颗粒流动性好,填充的重量更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0394]
实施例3方法1(酸性辅料=富马酸)和实施例3方法2(酸性辅料=枸橼酸)制备得到的胶囊内容物,胶囊内容物的装量差异
±
2.5%,低于限度标准
±
5%。
[0395]
实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸)制备得到的胶囊内容物,胶囊内容物的装量差异分别为
±
2.8%和
±
3.0%,虽然低于限度标准
±
5%。
[0396]
上述结果表明,实施例1(酸性辅料=酒石酸)、实施例3方法1(酸性辅料=富马酸)、实施例3方法2(酸性辅料=枸橼酸)、实施例3方法3(酸性辅料=乳酸)和实施例3方法4(酸性辅料=醋酸)制备得到的胶囊剂,其内容物的装量差异均符合限度标准。
[0397]
小结:
[0398]
①
采用本发明的酸性辅料(例如,枸橼酸、富马酸、酒石酸)能制备稳定性和溶出行为良好的胶囊剂;
[0399]
例如,实施例1酒石酸、实施例3方法1富马酸和实施例3方法2枸橼酸制备得到的胶囊剂,稳定性和溶出行为良好,具体表现为:
[0400]
所述胶囊剂中各杂质含量显著降低(0.005~0.05%,显著低于杂质限度标准≤0.1%),稳定性良好;溶出度高达98~100%,显著高于限度标准≥85%,崩解速度快(30s),溶出行为显著。
[0401]
②
其中,当采用的酸性辅料为酒石酸时,制备得到的胶囊剂(例如,实施例1),制备的胶囊剂稳定性和溶出行为最优,具体表现为:
[0402]
所述胶囊剂中各杂质含量低至0.005%,显著低于杂质限度标准≤0.1%,稳定性良好;溶出度高达100%,显著高于限度标准≥85%,崩解速度快(30s),溶出行为最显著。
[0403]
③
反之,采用本发明以外的酸性填料(例如,实施例3方法3乳酸和实施例3方法4醋酸)得到的胶囊剂稳定性和溶出行为差,无法得到合格的胶囊剂,具体表现为:
[0404]
所述胶囊剂的其他最大单杂含量高达0.39%,不符合限度标准(≤0.1%);稳定性差;崩解过程出现堆积,并且溶出度下降至81~83%,远低于限度标准≥85%,无法制备合格的胶囊剂。
[0405]
(三)结果分析(二)探究了酸性辅料的种类对胶囊剂质量的影响,以下内容通过改变酸性辅料酒石酸的用量探究了胶囊剂囊芯材料中酸性辅料酒石酸的不同用量对胶囊剂质量的影响:
[0406]
1、区别
[0407]
表19
[0408][0409]
2、制剂对比
[0410]
表20
[0411]
[0412][0413]
表21
[0414][0415]
表22
[0416][0417]
表23
[0418][0419]
[0420]
结论:
[0421]
1)杂质含量结果显示:
[0422]
新鲜制备条件下(0天):
[0423]
实施例1(酒石酸=2.0wt%)、实施例4方法1(酒石酸=0.3wt%)和实施例4方法2(酒石酸=10.0wt%)的pm4含量为0.006~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.007%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.10~0.15%,远低于限度标准(≤0.5%),稳定性良好。
[0424]
实施例4方法3(酒石酸=0.1wt%)的pm4含量为0.01%;杂质19含量为0.02%;其他最大单杂含量为0.07%,总杂含量为0.18%。
[0425]
实施例4方法4(酒石酸=15.0wt%)的pm4含量为0.01%;杂质19含量为0.02%;其他最大单杂含量为0.08%;总杂含量为0.13%;杂质含量高于实施例1(酒石酸=2.0wt%)的胶囊剂产品。
[0426]
高温40℃条件下放置30天:
[0427]
实施例1(酒石酸=2.0wt%)、实施例4方法1(酒石酸=0.3wt%)和实施例4方法2(酒石酸=10.0wt%)的pm4含量为0.007~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.01%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.08%,低于限度标准(≤0.1%);总杂含量为0.11~0.25%,均低于限度标准(≤0.5%),稳定性良好。
[0428]
实施例4方法3(酒石酸=0.1wt%)的pm4含量为0.02%,低于限度标准(≤0.1%);杂质19含量为0.05%,低于限度标准(≤0.1%);总杂含量为0.32%,低于限度标准(≤0.5%);其他最大单杂含量显著增加至0.16%,远高于限度标准(≤0.1%),无法制备稳定性合格的胶囊剂。
[0429]
实施例4方法4(酒石酸=15.0wt%)的pm4含量为0.02%;杂质19含量为0.04%;总杂含量为0.48%,符合限度标准(≤0.5%)。其他最大单杂含量显著增加至0.32%,不符合限度标准(≤0.1%);稳定性差,无法制备合格的胶囊剂产品。
[0430]
高湿75%条件下放置30天:
[0431]
实施例1(酒石酸=2.0wt%)、实施例4方法1(酒石酸=0.3wt%)和实施例4方法2(酒石酸=10.0wt%)的pm4含量为0.006~0.008%,低于限度标准(≤0.1%);杂质19含量为0.005~0.008%,低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.08%,低于限度标准(≤0.1%);总杂含量为0.12~0.20%,均低于限度标准(≤0.5%),稳定性良好。
[0432]
实施例4方法3(酒石酸=0.1wt%)的pm4含量为0.01%;杂质19含量为0.03%;总杂含量为0.28%。但是,其他最大单杂含量显著增加至0.15%,不符合限度标准(≤0.1%);稳定性差,无法制备合格的胶囊剂产品。
[0433]
实施例4方法4(酒石酸=15.0wt%)的pm4含量为0.01%;杂质19含量为0.03%。总杂含量增加至0.41%,远高于实施例1(酒石酸=2.0wt%)总杂含量;其他最大单杂含量显著增加至0.29%,不符合限度标准(≤0.1%);稳定性差,无法制备合格的胶囊剂产品。
[0434]
光照4500
±
500lx条件下放置30天:
[0435]
实施例1(酒石酸=2.0wt%)、实施例4方法1(酒石酸=0.3wt%)和实施例4方法2(酒石酸=10.0wt%)的pm4含量为0.006~0.008%,低于限度标准(≤0.1%);杂质19含量
为0.006~0.009%,低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.09%,低于限度标准(≤0.1%);总杂含量为0.13~0.23%,均低于限度标准(≤0.5%),稳定性良好。
[0436]
实施例4方法3(酒石酸=0.1wt%)的pm4含量为0.02%;杂质19含量为0.06%。总杂含量增加至0.31%,远高于实施例1(酒石酸=2.0wt%)总杂含量;其他最大单杂含量显著增加至0.19%,不符合限度标准(≤0.1%);稳定性差,无法制备合格的胶囊剂产品。
[0437]
实施例4方法4(酒石酸=15.0wt%)的pm4含量为0.02%;杂质19含量为0.03%。总杂含量增加至0.47%,远高于实施例1(酒石酸=2.0wt%)总杂含量;其他最大单杂含量显著增加至0.36%,不符合限度标准(≤0.1%);稳定性差,无法制备合格的胶囊剂产品。
[0438]
上述结果表明,实施例1(酒石酸=2.0wt%)制备得到的胶囊剂,其稳定性显著优于实施例4方法1(酒石酸=0.3wt%)、实施例4方法2(酒石酸=10.0wt%)、实施例4方法3(酒石酸=0.1wt%)和实施例4方法4(酒石酸=15.0wt%);并且实施例4方法4(酒石酸=15.0wt%)无法制备稳定性合格的胶囊剂产品。
[0439]
2)溶出行为结果显示:
[0440]
实施例1(酒石酸=2.0wt%)得到的制剂在30s完全崩散,杯底无堆积,溶出度为100%,远高于溶出度限度(≥85%),溶出行为良好且显著。
[0441]
实施例4方法1(酸性辅料=富马酸)、实施例4方法2(酸性辅料=枸橼酸)和实施例4方法4(酸性辅料=醋酸)得到的制剂在30s完全崩散,杯底无堆积,溶出度为90~98%,高于溶出度限度(≥85%),溶出行为良好。
[0442]
实施例4方法3(酸性辅料=乳酸)得到的制剂崩散时间延长至2min,并且杯底出现堆积,溶出度降低至83%,不符合溶出度限度标准(≥85%),溶出行为差,无法制备溶出行为合格的胶囊剂产品。
[0443]
上述结果表明,实施例1(酒石酸=2.0wt%)制备得到的胶囊剂,其溶出行为显著优于实施例4方法1(酒石酸=0.3wt%)、实施例4方法2(酒石酸=10.0wt%)、实施例4方法3(酒石酸=0.1wt%)和实施例4方法4(酒石酸=15.0wt%);并且实施例4方法3(酒石酸=0.1wt%)无法制备溶出行为合格的胶囊剂产品。
[0444]
3)胶囊剂内容物溶解过程中微环境ph结果显示:
[0445]
实施例1(酒石酸=2.0wt%)的胶囊内容物,在溶解过程中微环境ph为3.04。实施例4方法1(酒石酸=0.3wt%)和实施例4方法2(酒石酸=10.0wt%)微环境ph分别为4.12和3.04;微环境ph呈酸性条件,使得药物活性成分更易溶,进而显著提高胶囊剂的溶出度,并保持良好的稳定性。
[0446]
但是,实施例4方法3(酒石酸=0.1wt%)的微环境ph为4.62,酸性辅料用量过少,ph较实施例1(酒石酸=2.0wt%)显著升高,无法使药物活性成分充分的溶出,从而使溶出度下降。
[0447]
实施例4方法4(酒石酸=15.0wt%)的微环境ph为2.84,酸性辅料用量过多,由于自身易发生吸湿潮解,导致有关物质的杂质含量增加,降低了胶囊剂的稳定性。
[0448]
上述结果与1)杂质含量结果和2)溶出行为结果的高度一致,表明上述结果的准确性。
[0449]
4)胶囊填装结果显示:
[0450]
实施例1(酒石酸=2.0wt%)制备得到的胶囊内容物,颗粒流动性好,填充的重量
更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0451]
实施例4方法1(酒石酸=0.3wt%)、实施例4方法2(酒石酸=10.0wt%)、实施例4方法3(酒石酸=0.1wt%)和实施例4方法4(酒石酸=15.0wt%),制备得到的胶囊内容物,胶囊内容物的装量差异度升高至
±
2.3~3.9%,控制在限度标准
±
5%范围内。
[0452]
上述结果表明,实施例1(酒石酸=2.0wt%)制备得到的胶囊剂,其内容物的装量差异优于实施例4方法1(酒石酸=0.3wt%)、实施例4方法2(酒石酸=10.0wt%)、实施例4方法3(酒石酸=0.1wt%)和实施例4方法4(酒石酸=15.0wt%)。
[0453]
小结:
[0454]
①
采用本发明用量的酸性辅料(0.3~10wt%),能制备稳定性和溶出行为良好胶囊剂;
[0455]
例如,采用实施例1(酒石酸=2.0wt%)、实施例4方法1(酒石酸=0.3wt%)和实施例4方法2(酒石酸=10.0wt%)的酒石酸用量,制备得到的胶囊剂稳定性和溶出行为良好,具体表现为:
[0456]
所述胶囊剂中各杂质含量显著降低(0.005~0.09%,低于杂质限度标准≤0.1%),稳定性良好;溶出度为90~100%,高于限度标准≥85%,崩解速度快(30s),溶出行为显著。
[0457]
②
其中,当采用酸性辅料酒石酸用量为2.0wt%时,制备得到的胶囊剂(例如,实施例1),制备的胶囊剂稳定性和溶出行为最优,具体表现为:
[0458]
所述胶囊剂中各杂质含量低至0.005%,显著低于杂质限度标准≤0.1%,稳定性良好;溶出度高达100%,显著高于限度标准≥85%,崩解速度快(30s),溶出行为最显著。
[0459]
③
反之,采用本发明以外用量的酸性填料(<0.3wt%或>10wt%)得到的胶囊剂稳定性和溶出行为差,无法得到合格的胶囊剂;
[0460]
例如,采用实施例4方法3酒石酸=0.1wt%和实施例4方法4酒石酸=15.0wt%的酒石酸用量,得到的胶囊剂稳定性和溶出行为差,无法得到合格的胶囊剂,具体表现为:
[0461]
所述胶囊剂的其他最大单杂含量高达0.36%,不符合限度标准(≤0.1%);稳定性差;崩解时间延长至2min,崩解过程出现堆积,并且溶出度下降至83%,远低于限度标准≥85%,无法制备合格的胶囊剂。
[0462]
(四)为了探究崩解剂用量对胶囊剂质量的影响,改变囊芯材料中崩解剂交联羧甲基纤维素钠的质量百分含量:
[0463]
1、区别
[0464]
表24
[0465]
实施例崩解剂-交联羧甲基纤维素钠的质量百分含量/wt%实施例15.0实施例5方法12.0实施例5方法28.0实施例5方法31.5
[0466]
2、制剂对比
[0467]
表25
[0468][0469]
表26
[0470][0471]
表27
[0472][0473]
结论:
[0474]
1)杂质含量结果显示:
[0475]
新鲜制备条件下(0天):
[0476]
实施例1(交联羧甲基纤维素钠=5.0wt%)以及实施例5方法1(交联羧甲基纤维素钠=2.0wt%)、实施例5方法2(交联羧甲基纤维素钠=8.0wt%)和实施例5方法3(交联羧甲基纤维素钠=1.5wt%)的pm4含量为0.006~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.007%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.10~0.12%,远低于限度标准(≤0.5%),稳定性
良好。
[0477]
高温40℃条件下放置30天:
[0478]
实施例1(交联羧甲基纤维素钠=5.0wt%)以及实施例5方法1(交联羧甲基纤维素钠=2.0wt%)、实施例5方法2(交联羧甲基纤维素钠=8.0wt%)和实施例5方法3(交联羧甲基纤维素钠=1.5wt%)的pm4含量为0.007~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.008%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.07%,远低于限度标准(≤0.1%);总杂含量为0.11~0.16%,远低于限度标准(≤0.5%),稳定性良好。
[0479]
高湿75%条件下放置30天:
[0480]
实施例1(交联羧甲基纤维素钠=5.0wt%)以及实施例5方法1(交联羧甲基纤维素钠=2.0wt%)、实施例5方法2(交联羧甲基纤维素钠=8.0wt%)和实施例5方法3(交联羧甲基纤维素钠=1.5wt%)的pm4含量为0.007~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.007~0.008%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.12~0.14%,远低于限度标准(≤0.5%),稳定性良好。
[0481]
光照4500
±
500lx条件下放置30天:
[0482]
实施例1(交联羧甲基纤维素钠=5.0wt%)以及实施例5方法1(交联羧甲基纤维素钠=2.0wt%)、实施例5方法2(交联羧甲基纤维素钠=8.0wt%)和实施例5方法3(交联羧甲基纤维素钠=1.5wt%)的pm4含量为0.008~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.007~0.01%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.06%,远低于限度标准(≤0.1%);总杂含量为0.13~0.15%,远低于限度标准(≤0.5%),稳定性良好。
[0483]
2)溶出行为结果显示:
[0484]
实施例1(交联羧甲基纤维素钠=5.0wt%)得到的制剂在30s完全崩散,杯底无堆积,溶出度为100%,远高于溶出度限度(≥85%),溶出行为良好且显著。
[0485]
实施例5方法2(交联羧甲基纤维素钠=8.0wt%)得到的制剂在30s完全崩散,杯底无堆积,溶出度为98%,溶出度高于溶出度限度(≥85%),溶出行为良好。
[0486]
实施例5方法1(交联羧甲基纤维素钠=2.0wt%)得到的制剂完全崩散时间延长至1min,杯底无堆积,溶出度为90%,溶出度高于溶出度限度(≥85%),溶出行为符合标准。
[0487]
实施例5方法3(交联羧甲基纤维素钠=1.5wt%)得到的制剂崩散时间延长至2min,并且杯底出现堆积,溶出度降低至80%,不符合溶出度限度标准(≥85%),溶出行为差,无法制备溶出行为合格的胶囊剂产品。
[0488]
上述结果表明,实施例1(交联羧甲基纤维素钠=5.0wt%)制备得到的胶囊剂,其溶出行为优于实施例5方法1(交联羧甲基纤维素钠=2.0wt%)和实施例5方法2(交联羧甲基纤维素钠=8.0wt%);实施例5方法3(交联羧甲基纤维素钠=1.5wt%)无法制备溶出行为合格的胶囊剂产品。
[0489]
3)胶囊填装结果显示:
[0490]
实施例1(交联羧甲基纤维素钠=5.0wt%)制备得到的胶囊内容物,颗粒流动性好,填充的重量更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0491]
实施例5方法1(交联羧甲基纤维素钠=2.0wt%)、实施例5方法2(交联羧甲基纤维素钠=8.0wt%)、实施例5方法3(交联羧甲基纤维素钠=1.5wt%)制备得到的胶囊内容物,胶囊内容物的装量差异度升高至
±
2.5~2.8%,控制在限度标准
±
5%范围内。
[0492]
上述结果表明,实施例1(交联羧甲基纤维素钠=5.0wt%)制备得到的胶囊剂,其内容物的装量差异优于实施例5方法1(交联羧甲基纤维素钠=2.0wt%)、实施例5方法2(交联羧甲基纤维素钠=8.0wt%)和实施例5方法3(交联羧甲基纤维素钠=1.5wt%)。
[0493]
小结:
[0494]
①
采用本发明用量的崩解剂(2.0~8.0wt%)能制备溶出行为良好的胶囊剂;
[0495]
例如,实施例1(交联羧甲基纤维素钠=5.0wt%)、实施例5方法1(交联羧甲基纤维素钠=2.0wt%)和实施例5方法2(交联羧甲基纤维素钠=8.0wt%)用量的崩解剂,制备的胶囊剂溶出行为良好,具体表现为:
[0496]
所述胶囊剂的溶出度为90~100%,高于限度标准≥85%,崩解速度快(30s),溶出行为显著。
[0497]
②
其中,当采用崩解剂交联羧甲基纤维素钠的用量为5.0wt%时,制备得到的胶囊剂(例如,实施例1)溶出行为最优;具体表现为:
[0498]
所述胶囊剂的溶出度高达100%,显著高于限度标准≥85%,崩解速度快(30s),溶出行为最显著。
[0499]
③
反之,采用本发明以外用量崩解剂(<2.0wt%或>8.0wt%)得到的胶囊剂溶出行为差,无法得到合格的胶囊剂;
[0500]
例如,采用实施例5方法3交联羧甲基纤维素钠=1.5wt%用量的崩解剂,得到的胶囊剂溶出行为差,无法得到合格的胶囊剂,具体表现为:
[0501]
所述胶囊剂的崩解时间延长至2min,崩解过程出现堆积,并且溶出度下降至80%,远低于限度标准≥85%,无法制备合格的胶囊剂。
[0502]
(五)为了探究崩解剂的种类对胶囊剂质量的影响,改变崩解剂的种类:
[0503]
1、区别
[0504]
表28
[0505]
实施例崩解剂实施例1交联羧甲基纤维素钠实施例6方法1低取代羟丙基纤维素实施例6方法2羧甲基淀粉钠实施例6方法3交联聚维酮实施例6方法4交联羧甲基纤维素钠和交联聚维酮
[0506]
2、制剂对比
[0507]
表29
[0508][0509]
表30
[0510][0511]
表31
[0512][0513][0514]
结论:
[0515]
1)杂质含量结果显示:
[0516]
新鲜制备条件下(0天):
[0517]
实施例1(崩解剂=交联羧甲基纤维素钠)以及实施例6方法1(崩解剂=低取代羟丙基纤维素)、实施例6方法2(崩解剂=羧甲基淀粉钠)、实施例6方法3(崩解剂=交联聚维酮)、实施例6方法4(崩解剂=交联羧甲基纤维素钠+交联聚维酮)的pm4含量为0.006~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.006%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.10~0.12%,远低于限度标准(≤0.5%),稳定性良好。
[0518]
高温40℃条件下放置30天:
[0519]
实施例1(崩解剂=交联羧甲基纤维素钠)以及实施例6方法1(崩解剂=低取代羟丙基纤维素)、实施例6方法2(崩解剂=羧甲基淀粉钠)、实施例6方法3(崩解剂=交联聚维酮)、实施例6方法4(崩解剂=交联羧甲基纤维素钠+交联聚维酮)的pm4含量为0.007~0.009%,远低于限度标准(≤0.1%);杂质19含量均为0.006%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.06%,远低于限度标准(≤0.1%);总杂含量为0.11~0.15%,远低于限度标准(≤0.5%),稳定性良好。
[0520]
高湿75%条件下放置30天:
[0521]
实施例1(崩解剂=交联羧甲基纤维素钠)以及实施例6方法1(崩解剂=低取代羟丙基纤维素)、实施例6方法2(崩解剂=羧甲基淀粉钠)、实施例6方法3(崩解剂=交联聚维酮)、实施例6方法4(崩解剂=交联羧甲基纤维素钠+交联聚维酮)的pm4含量为0.006~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.008%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.12~0.14%,远低于限度标准(≤0.5%),稳定性良好。
[0522]
光照4500
±
500lx条件下放置30天:
[0523]
实施例1(崩解剂=交联羧甲基纤维素钠)以及实施例6方法1(崩解剂=低取代羟丙基纤维素)、实施例6方法2(崩解剂=羧甲基淀粉钠)、实施例6方法3(崩解剂=交联聚维酮)、实施例6方法4(崩解剂=交联羧甲基纤维素钠+交联聚维酮)的pm4含量为0.006~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.008%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.12~0.13%,远低于限度标准(≤0.5%),稳定性良好。
[0524]
上述结果表明,采用本发明的崩解剂,均能得到稳定性良好的胶囊剂产品。
[0525]
2)溶出行为结果显示:
[0526]
实施例1(崩解剂=交联羧甲基纤维素钠)得到的制剂在30s完全崩散,杯底无堆积,溶出度为100%,远高于溶出度限度(≥85%),溶出行为良好且显著。
[0527]
实施例6方法1(崩解剂=低取代羟丙基纤维素)、实施例6方法2(崩解剂=羧甲基淀粉钠)、实施例6方法3(崩解剂=交联聚维酮)、实施例6方法4(崩解剂=交联羧甲基纤维素钠+交联聚维酮)得到的制剂在30s完全崩散,杯底无堆积,溶出度为96~97%,较实施例1(崩解剂=交联羧甲基纤维素钠)溶解度略有降低,但溶出度仍显著高于溶出度限度(≥85%),溶出行为良好。
[0528]
上述结果表明,采用本发明的崩解剂,均能得到溶出行为良好的胶囊剂产品。
[0529]
3)胶囊填装结果显示:
[0530]
实施例1(崩解剂=交联羧甲基纤维素钠)制备得到的胶囊内容物,颗粒流动性好,填充的重量更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0531]
实施例6方法1(崩解剂=低取代羟丙基纤维素)、实施例6方法2(崩解剂=羧甲基淀粉钠)、实施例6方法3(崩解剂=交联聚维酮)、实施例6方法4(崩解剂=交联羧甲基纤维素钠+交联聚维酮)制备得到的胶囊内容物,胶囊内容物的装量差异度控制在
±
3.0~3.2%,略有升高,但仍控制在限度标准
±
5%范围内。
[0532]
上述结果表明,采用本发明的崩解剂,均能得到内容物的装量差异优异且符合限度标准的胶囊剂产品。
[0533]
小结:
[0534]
①
采用本发明的崩解剂制备得到的胶囊剂(例如,羧甲基淀粉钠、交联羧甲基纤维素钠、低取代羟丙基纤维素、交联聚维酮中的任意一种或几种),能制备稳定性和溶出行为良好,装量差异小、均匀度高的胶囊剂;
[0535]
例如,采用实施例1(崩解剂=交联羧甲基纤维素钠)、实施例6方法1(崩解剂=低取代羟丙基纤维素)、实施例6方法2(崩解剂=羧甲基淀粉钠)、实施例6方法3(崩解剂=交联聚维酮)、实施例6方法4(崩解剂=交联羧甲基纤维素钠+交联聚维酮)上述崩解剂种类,制备的胶囊剂稳定性和溶出行为良好,并且装量差异小、均匀度高,具体表现为:
[0536]
所述胶囊剂中各杂质含量显著降低(0.005~0.06%,显著低于杂质限度标准≤0.1%),稳定性良好;溶出度为96~100%,高于限度标准≥85%,崩解速度快(30s),溶出行为显著;并且胶囊剂内容物的装量差异
±
1.8~3.2%,远小于限度标准
±
5%,均匀度良好;
[0537]
②
其中,当采用崩解剂为交联羧甲基纤维素钠时,制备得到的胶囊剂(例如,实施例1),制备的胶囊剂稳定性和溶出行为最优,装量差异最小、均匀度最高;具体表现为:
[0538]
所述胶囊剂中各杂质含量低至0.005%,显著低于杂质限度标准≤0.1%,稳定性良好;溶出度高达100%,显著高于限度标准≥85%,崩解速度快(30s),溶出行为最显著;并且胶囊剂内容物的装量差异低至
±
1.8%,远小于限度标准
±
5%,均匀度良好。
[0539]
(六)、为了探究填充剂的不同用量对胶囊剂质量的影响,改变囊芯材料中填充剂微晶纤维素的质量百分含量;
[0540]
1、区别
[0541]
表32
[0542]
实施例填充剂-微晶纤维素的质量百分含量/wt%实施例120实施例7方法115实施例7方法250实施例7方法310实施例7方法455
[0543]
2、制剂对比
[0544]
表33
[0545][0546]
表34
[0547][0548]
表35
[0549][0550][0551]
结论:
[0552]
1)杂质含量结果显示:
[0553]
新鲜制备条件下(0天):
[0554]
实施例1(微晶纤维素=20wt%)以及实施例7方法1(微晶纤维素=15wt%)、实施例7方法2(微晶纤维素=50wt%)、实施例7方法3(微晶纤维素=10wt%)和实施例7方法4(微晶纤维素=55wt%)的pm4含量为0.006~0.007%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.007%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.10~0.12%,远低于限度标准(≤0.5%),稳定性良好。
[0555]
高温40℃条件下放置30天:
[0556]
实施例1(微晶纤维素=20wt%)以及实施例7方法1(微晶纤维素=15wt%)、实施例7方法2(微晶纤维素=50wt%)、实施例7方法3(微晶纤维素=10wt%)和实施例7方法4(微晶纤维素=55wt%)的pm4含量为0.007~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.008%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.07%,远低于限度标准(≤0.1%);总杂含量为0.11~0.16%,远低于限度标准(≤0.5%),稳定性良好。
[0557]
高湿75%条件下放置30天:
[0558]
实施例1(微晶纤维素=20wt%)以及实施例7方法1(微晶纤维素=15wt%)、实施例7方法2(微晶纤维素=50wt%)、实施例7方法3(微晶纤维素=10wt%)和实施例7方法4(微晶纤维素=55wt%)的pm4含量为0.007~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.008%,远低于限度标准(≤0.1%);其他最大单杂含量均为0.05%,远低于限度标准(≤0.1%);总杂含量为0.12~0.14%,远低于限度标准(≤0.5%),稳定性良好。
[0559]
光照4500
±
500lx条件下放置30天:
[0560]
实施例1(微晶纤维素=20wt%)以及实施例7方法1(微晶纤维素=15wt%)、实施例7方法2(微晶纤维素=50wt%)、实施例7方法3(微晶纤维素=10wt%)和实施例7方法4(微晶纤维素=55wt%)的pm4含量为0.006~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.01%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.06%,远低于限度标准(≤0.1%);总杂含量为0.12~0.15%,远低于限度标准(≤0.5%),稳定性良好。
[0561]
2)溶出行为结果显示:
[0562]
实施例1(交联羧甲基纤维素钠=5.0wt%)得到的制剂在30s完全崩散,杯底无堆积,溶出度为100%,远高于溶出度限度(≥85%),溶出行为良好且显著。
[0563]
实施例7方法1(微晶纤维素=15wt%)、实施例7方法2(微晶纤维素=50wt%)、实施例7方法3(微晶纤维素=10wt%)得到的制剂在30s完全崩散,杯底无堆积,溶出度为96~97%,溶出度高于溶出度限度(≥85%),溶出行为良好。
[0564]
实施例7方法4(微晶纤维素=55wt%)得到的制剂崩散过程杯底出现堆积,溶出度降低至82%,不符合溶出度限度标准(≥85%),溶出行为差,无法制备溶出行为合格的胶囊剂产品。
[0565]
上述结果表明,实施例1(交联羧甲基纤维素钠=5.0wt%)制备得到的胶囊剂,其溶出行为优于实施例7方法1(微晶纤维素=15wt%)、实施例7方法2(微晶纤维素=
50wt%)、实施例7方法3(微晶纤维素=10wt%);实施例7方法4(微晶纤维素=55wt%)无法制备溶出行为合格的胶囊剂产品。
[0566]
3)胶囊填装结果显示:
[0567]
实施例1(微晶纤维素=20wt%)制备得到的胶囊内容物,颗粒流动性好,填充的重量更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0568]
实施例7方法1(微晶纤维素=15wt%)、实施例7方法2(微晶纤维素=50wt%)和实施例7方法4(微晶纤维素=55wt%)制备得到的胶囊内容物,胶囊内容物的装量差异为
±
2.3~4.2%,控制在限度标准
±
5%范围内。
[0569]
但实施例7方法3(微晶纤维素=10wt%)制备得到的胶囊内容物,胶囊内容物的装量差异度升高至
±
5.8%,显著高于限度标准
±
5%范围,无法得到装量差异合格的胶囊剂产品。
[0570]
上述结果表明,实施例7方法1(微晶纤维素=15wt%)制备得到的胶囊剂,其内容物的装量差异优于实施例7方法1(微晶纤维素=15wt%)、实施例7方法2(微晶纤维素=50wt%)和实施例7方法4(微晶纤维素=55wt%);实施例7方法3(微晶纤维素=10wt%)无法得到装量差异合格的胶囊剂产品。
[0571]
小结:
[0572]
①
采用本发明用量的填充剂(15~50wt%),能制备溶出行为良好、装量差异小、均匀度高的胶囊剂;
[0573]
例如,采用实施例1(微晶纤维素=20wt%)、实施例7方法1(微晶纤维素=15wt%)和实施例7方法2(微晶纤维素=50wt%)上述用量,制备得到的胶囊剂溶出行为良好、装量差异小、、均匀度高,具体表现为:
[0574]
所述胶囊剂的溶出度为96~100%,高于限度标准≥85%,崩解速度快(30s),溶出行为显著;并且胶囊剂内容物的装量差异
±
1.8~4.2%,远小于限度标准
±
5%,均匀度高。
[0575]
②
其中,当采用填充剂微晶纤维素的用量为20wt%时,制备得到的胶囊剂(例如,实施例1),制备的胶囊剂溶出行为最优、装量差异最小,均匀度最高;具体表现为:
[0576]
所述胶囊剂的溶出度高达100%,显著高于限度标准≥85%,崩解速度快(30s),溶出行为最显著;并且胶囊剂内容物的装量差异低至
±
1.8%,远小于限度标准
±
5%,均匀度最高;
[0577]
③
反之,采用本发明以外用量填充剂(<15wt%或50wt%)得到的胶囊剂溶出行为差、装量差异大,无法得到合格的胶囊剂;
[0578]
例如,实施例7方法3微晶纤维素=10wt%,实施例7方法4微晶纤维素=55wt%得到的胶囊剂装量差异大、溶出行为差,无法得到合格的胶囊剂,具体表现为:
[0579]
所述胶囊剂崩解过程出现堆积,并且溶出度下降至80%,远低于限度标准≥85%;并且胶囊剂的装量差异高达
±
5.8%,显著高于限度标准
±
5%范围;无法制备合格的胶囊剂。
[0580]
(七)为了探究填充剂的种类对胶囊剂质量的影响,改变填充剂的种类:
[0581]
1、区别
[0582]
表36
[0583][0584][0585]
2、制剂对比
[0586]
表37
[0587][0588]
表38
[0589]
[0590]
表39
[0591][0592][0593]
结论:
[0594]
1)杂质含量结果显示:
[0595]
新鲜制备条件下(0天):
[0596]
实施例1(填充剂=微晶纤维素)以及实施例8方法1(填充剂=乳糖)、实施例8方法2(填充剂=预胶化淀粉)、实施例8方法3(填充剂=碳酸氢钙)、实施例8方法4(填充剂=碳酸钙)的pm4含量为0.006~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.007%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.10~0.12%,远低于限度标准(≤0.5%),稳定性良好。
[0597]
高温40℃条件下放置30天:
[0598]
实施例1(填充剂=微晶纤维素)以及实施例8方法1(填充剂=乳糖)、实施例8方法2(填充剂=预胶化淀粉)、实施例8方法3(填充剂=碳酸氢钙)、实施例8方法4(填充剂=碳酸钙)的pm4含量为0.007~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.009%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.07%,远低于限度标准(≤0.1%);总杂含量为0.11~0.16%,远低于限度标准(≤0.5%),稳定性良好。
[0599]
高湿75%条件下放置30天:
[0600]
实施例1(填充剂=微晶纤维素)以及实施例8方法1(填充剂=乳糖)、实施例8方法2(填充剂=预胶化淀粉)、实施例8方法3(填充剂=碳酸氢钙)、实施例8方法4(填充剂=碳酸钙)的pm4含量为0.007~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.01%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.06%,远低于限度标准(≤0.1%);总杂含量为0.12~0.14%,远低于限度标准(≤0.5%),稳定性良好。
[0601]
光照4500
±
500lx条件下放置30天:
[0602]
实施例1(填充剂=微晶纤维素)以及实施例8方法1(填充剂=乳糖)、实施例8方法2(填充剂=预胶化淀粉)、实施例8方法3(填充剂=碳酸氢钙)、实施例8方法4(填充剂=碳酸钙)的pm4含量为0.008~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.008~0.01%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.06%,远低于限度标准(≤0.1%);总杂含量为0.13~0.15%,远低于限度标准(≤0.5%),稳定性良好。
[0603]
上述结果表明,采用本发明的填充剂,均能得到稳定性良好的胶囊剂产品。
[0604]
2)溶出行为结果显示:
[0605]
实施例1(填充剂=微晶纤维素)得到的制剂在30s完全崩散,杯底无堆积,溶出度
为100%,远高于溶出度限度(≥85%),溶出行为良好且显著。
[0606]
实施例8方法1(填充剂=乳糖)、实施例8方法2(填充剂=预胶化淀粉)、实施例8方法3(填充剂=碳酸氢钙)、实施例8方法4(填充剂=碳酸钙)得到的制剂在30s完全崩散,杯底无堆积,溶出度为97~98%,较实施例1(填充剂=交联羧甲基纤维素钠)溶解度略有降低,但溶出度仍显著高于溶出度限度(≥85%),溶出行为良好。
[0607]
上述结果表明,采用本发明的填充剂,均能得到溶出行为良好的胶囊剂产品。
[0608]
3)胶囊填装结果显示:
[0609]
实施例1(填充剂=微晶纤维素)制备得到的胶囊内容物,颗粒流动性好,填充的重量更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0610]
实施例8方法1(填充剂=乳糖)、实施例8方法2(填充剂=预胶化淀粉)、实施例8方法3(填充剂=碳酸氢钙)、实施例8方法4(填充剂=碳酸钙)制备得到的胶囊内容物,胶囊内容物的装量差异度控制在
±
2.9~3.6%,略有升高,但仍控制在限度标准
±
5%范围内。
[0611]
上述结果表明,采用本发明的填充剂,均能得到内容物的装量差异优异且符合限度标准的胶囊剂产品。
[0612]
小结:
[0613]
①
采用本发明的填充剂(乳糖、微晶纤维素、预胶化淀粉、磷酸氢钙、碳酸钙中的任意一种或几种),能制备得到稳定性和溶出行为良好,装量差异小、均匀度高的胶囊剂。
[0614]
例如,实施例1(填充剂=微晶纤维素)、实施例8方法1(填充剂=乳糖)、实施例8方法2(填充剂=预胶化淀粉)、实施例8方法3(填充剂=碳酸氢钙)、实施例8方法4(填充剂=碳酸钙),制备得到的胶囊剂稳定性和溶出行为良好,装量差异小、均匀度高,具体表现为:
[0615]
所述胶囊剂中各杂质含量显著降低(0.005~0.07%,显著低于杂质限度标准≤0.1%),稳定性良好;溶出度为97~100%,高于限度标准≥85%,崩解速度快(30s),溶出行为显著;并且胶囊剂内容物的装量差异
±
1.8~3.6%,小于限度标准
±
5%,均匀度良好。
[0616]
②
其中,当采用填充剂为微晶纤维素时,制备得到的胶囊剂(例如,实施例1),制备的胶囊剂稳定性和溶出行为最优,装量差异最小、均匀度最高;具体表现为:
[0617]
所述胶囊剂中各杂质含量低至0.005%,显著低于杂质限度标准≤0.1%,稳定性良好;溶出度高达100%,显著高于限度标准≥85%,崩解速度快(30s),溶出行为最显著;并且胶囊剂内容物的装量差异低至
±
1.8%,远小于限度标准
±
5%,均匀度最高。
[0618]
(八)、为了探究润滑剂的不同用量对胶囊剂质量的影响,改变囊芯材料中润滑剂硬脂酸镁的质量百分含量;
[0619]
1、区别
[0620]
表40
[0621]
实施例润滑剂-硬脂酸镁的质量百分含量/wt%实施例11.0实施例9方法10.5实施例9方法22.0实施例9方法30.3实施例9方法42.5
[0622]
2、制剂对比
[0623]
表41
[0624][0625][0626]
表42
[0627][0628]
表43
[0629][0630]
结论:
[0631]
1)杂质含量结果显示:
[0632]
新鲜制备条件下(0天):
[0633]
实施例1(硬脂酸镁=1.0wt%)以及实施例9方法1(硬脂酸镁=0.5wt%)、实施例9方法2(硬脂酸镁=2.0wt%)、实施例9方法3(硬脂酸镁=0.3wt%)和实施例9方法4(硬脂酸镁=2.5wt%)的pm4含量为0.006~0.007%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.007%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.10~0.12%,远低于限度标准(≤0.5%),稳定性良好。
[0634]
高温40℃条件下放置30天:
[0635]
实施例1(硬脂酸镁=1.0wt%)以及实施例9方法1(硬脂酸镁=0.5wt%)、实施例9方法2(硬脂酸镁=2.0wt%)、实施例9方法3(硬脂酸镁=0.3wt%)和实施例9方法4(硬脂酸镁=2.5wt%)的pm4含量为0.007~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.008%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.07%,远低于限度标准(≤0.1%);总杂含量为0.11~0.16%,远低于限度标准(≤0.5%),稳定性良好。
[0636]
高湿75%条件下放置30天:
[0637]
实施例1(硬脂酸镁=1.0wt%)以及实施例9方法1(硬脂酸镁=0.5wt%)、实施例9方法2(硬脂酸镁=2.0wt%)、实施例9方法3(硬脂酸镁=0.3wt%)和实施例9方法4(硬脂酸镁=2.5wt%)的pm4含量为0.007~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.008%,远低于限度标准(≤0.1%);其他最大单杂含量均为0.05%,远低于限度标准(≤0.1%);总杂含量为0.12~0.14%,远低于限度标准(≤0.5%),稳定性良好。
[0638]
光照4500
±
500lx条件下放置30天:
[0639]
实施例1(硬脂酸镁=1.0wt%)以及实施例9方法1(硬脂酸镁=0.5wt%)、实施例9方法2(硬脂酸镁=2.0wt%)、实施例9方法3(硬脂酸镁=0.3wt%)和实施例9方法4(硬脂酸镁=2.5wt%)的pm4含量为0.006~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.01%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.06%,远低于限度标准(≤0.1%);总杂含量为0.12~0.15%,远低于限度标准(≤0.5%),稳定性良好。
[0640]
2)溶出行为结果显示:
[0641]
实施例1(硬脂酸镁=1.0wt%)得到的制剂在30s完全崩散,杯底无堆积,溶出度为100%,远高于溶出度限度(≥85%),溶出行为良好且显著。
[0642]
实施例9方法1(硬脂酸镁=0.5wt%)、实施例9方法2(硬脂酸镁=2.0wt%)和实施例9方法3(硬脂酸镁=0.3wt%)得到的制剂在30s~1min完全崩散,杯底无堆积,溶出度为97~98%,溶出度高于溶出度限度(≥85%),溶出行为良好。
[0643]
实施例9方法4(硬脂酸镁=2.5wt%)得到的制剂崩散时间延长至2min,显著增加为实施例1(硬脂酸镁=1.0wt%)崩散时间的4倍,并且崩散过程杯底出现堆积,溶出度降低至81%,不符合溶出度限度标准(≥85%),溶出行为差,无法制备溶出行为合格的胶囊剂产品。
[0644]
上述结果表明,实施例1(硬脂酸镁=1.0wt%)制备得到的胶囊剂,其溶出行为优于实施例9方法1(硬脂酸镁=0.5wt%)、实施例9方法2(硬脂酸镁=2.0wt%)和实施例9方法3(硬脂酸镁=0.3wt%);实施例9方法4(硬脂酸镁=2.5wt%)无法制备溶出行为合格的胶囊剂产品。
[0645]
3)胶囊填装结果显示:
[0646]
实施例1(硬脂酸镁=1.0wt%)制备得到的胶囊内容物,颗粒流动性好,填充的重量更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0647]
实施例9方法1(硬脂酸镁=0.5wt%)、实施例9方法2(硬脂酸镁=2.0wt%)和实施例9方法4(硬脂酸镁=2.5wt%)制备得到的胶囊内容物,胶囊内容物的装量差异为
±
2.5~4.3%,控制在限度标准
±
5%范围内。
[0648]
但实施例9方法3(硬脂酸镁=0.3wt%)制备得到的胶囊内容物,胶囊内容物的装量差异度升高至
±
5.9%,显著高于限度标准
±
5%范围,无法得到装量差异合格的胶囊剂产品。
[0649]
上述结果表明,实施例9方法1(硬脂酸镁=1.0wt%)制备得到的胶囊剂,其内容物的装量差异优于实施例9方法1(硬脂酸镁=0.5wt%)、实施例9方法2(硬脂酸镁=2.0wt%)和实施例9方法4(硬脂酸镁=2.5wt%);实施例9方法3(硬脂酸镁=0.3wt%)无法得到装量差异合格的胶囊剂产品。
[0650]
小结:
[0651]
①
采用本发明用量的润滑剂(0.5~2.0wt%)能制备溶出行为良好,装量差异小、均匀度高的胶囊剂;
[0652]
例如,采用实施例1(硬脂酸镁=1.0wt%)、实施例9方法1(硬脂酸镁=0.5wt%)和实施例9方法2(硬脂酸镁=2.0wt%)上述润滑剂用量,制备的胶囊剂溶出行为良好,并且装量差异小、均匀度高,具体表现为:
[0653]
所述胶囊剂的溶出度为97~100%,高于限度标准≥85%,崩解速度快(30s~1min),溶出行为显著;并且胶囊剂内容物的装量差异
±
1.8~4.3%,小于限度标准
±
5%,均匀度良好。
[0654]
②
其中,当采用润滑剂硬脂酸镁用量为1.0wt%时,制备得到的胶囊剂(例如,实施例1),制备的胶囊剂溶出行为最优;装量差异最小、均匀度最高;具体表现为:
[0655]
所述胶囊剂的溶出度高达100%,显著高于限度标准≥85%,崩解速度快(30s),溶出行为最显著;并且胶囊剂内容物的装量差异低至
±
1.8%,远小于限度标准
±
5%;均匀度最优。
[0656]
③
反之,采用本发明以外用量润滑剂(<0.5wt%或>2.0wt%)得到的胶囊剂溶出行为差,装量差异大、均匀度差;无法得到合格的胶囊剂;
[0657]
例如,实施例9方法3硬脂酸镁=0.3wt%,实施例9方法4硬脂酸镁=2.5wt%用量的润滑剂,得到的胶囊剂溶出行为差,装量差异大、均匀度差,无法得到合格的胶囊剂;具体表现为:
[0658]
所述胶囊剂崩散时间显著延长至2min,崩散过程出现堆积,并且溶出度下降至81%,远低于限度标准≥85%;并且胶囊剂内容物的装量差异高达
±
5.9%,显著高于限度标准
±
5%范围;无法制备合格的胶囊剂。
[0659]
(九)为了探究润滑剂的种类对胶囊剂质量的影响,改变润滑剂的种类:
[0660]
1、区别
[0661]
表44
[0662]
实施例润滑剂的种类
实施例1硬脂酸镁实施例10方法1胶态二氧化硅实施例10方法2硬脂酸钙实施例10方法3滑石粉
[0663]
2、制剂对比
[0664]
表45
[0665][0666][0667]
表46
[0668][0669]
表47
[0670][0671]
结论:
[0672]
1)杂质含量结果显示:
[0673]
新鲜制备条件下(0天):
[0674]
实施例1(润滑剂=硬脂酸镁)以及实施例10方法1(润滑剂=胶态二氧化硅)、实施例10方法2(润滑剂=硬脂酸钙)、实施例10方法3(润滑剂=滑石粉)的pm4含量为0.006~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.007%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.10~0.12%,远低于限度标准(≤0.5%),稳定性良好。
[0675]
高温40℃条件下放置30天:
[0676]
实施例1(润滑剂=硬脂酸镁)以及实施例10方法1(润滑剂=胶态二氧化硅)、实施例10方法2(润滑剂=硬脂酸钙)、实施例10方法3(润滑剂=滑石粉)的pm4含量为0.007~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.009%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.07%,远低于限度标准(≤0.1%);总杂含量为0.11~0.16%,远低于限度标准(≤0.5%),稳定性良好。
[0677]
高湿75%条件下放置30天:
[0678]
实施例1(润滑剂=硬脂酸镁)以及实施例10方法1(润滑剂=胶态二氧化硅)、实施例10方法2(润滑剂=硬脂酸钙)、实施例10方法3(润滑剂=滑石粉)的pm4含量为0.007~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.01%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.06%,远低于限度标准(≤0.1%);总杂含量为0.12~0.14%,远低于限度标准(≤0.5%),稳定性良好。
[0679]
光照4500
±
500lx条件下放置30天:
[0680]
实施例1(润滑剂=硬脂酸镁)以及实施例10方法1(润滑剂=胶态二氧化硅)、实施例10方法2(润滑剂=硬脂酸钙)、实施例10方法3(润滑剂=滑石粉)的pm4含量为0.008~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.008~0.01%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.06%,远低于限度标准(≤0.1%);总杂含量为0.13~0.15%,远低于限度标准(≤0.5%),稳定性良好。
[0681]
上述结果表明,采用本发明的润滑剂,均能得到稳定性良好的胶囊剂产品。
[0682]
2)溶出行为结果显示:
[0683]
实施例1(润滑剂=硬脂酸镁)得到的制剂在30s完全崩散,杯底无堆积,溶出度为100%,远高于溶出度限度(≥85%),溶出行为良好且显著。
[0684]
实施例10方法1(润滑剂=胶态二氧化硅)、实施例10方法2(润滑剂=硬脂酸钙)、实施例10方法3(润滑剂=滑石粉)得到的制剂在30s完全崩散,杯底无堆积,溶出度为96~97%,较实施例1(润滑剂=硬脂酸镁)溶解度略有降低,但溶出度仍显著高于溶出度限度(≥85%),溶出行为良好。
[0685]
上述结果表明,采用本发明的润滑剂,均能得到溶出行为良好的胶囊剂产品。
[0686]
3)胶囊填装结果显示:
[0687]
实施例1(润滑剂=硬脂酸镁)制备得到的胶囊内容物,颗粒流动性好,填充的重量更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0688]
实施例10方法1(润滑剂=胶态二氧化硅)、实施例10方法2(润滑剂=硬脂酸钙)、实施例10方法3(润滑剂=滑石粉)制备得到的胶囊内容物,胶囊内容物的装量差异度控制
在
±
2.5~2.8%,略有升高,但仍控制在限度标准
±
5%范围内。
[0689]
上述结果表明,采用本发明的润滑剂,均能得到内容物的装量差异优异且符合限度标准的胶囊剂产品。
[0690]
小结:
[0691]
①
采用本发明的润滑剂(硬脂酸镁、胶态二氧化硅、硬脂酸钙、滑石粉中的任意一种或几种)制备得到的胶囊剂稳定性和溶出行为良好,并且内容物的装量差异小、均匀度高;
[0692]
例如,采用实施例1(润滑剂=硬脂酸镁)、实施例10方法1(润滑剂=胶态二氧化硅)、实施例10方法2(润滑剂=硬脂酸钙)、实施例10方法3(润滑剂=滑石粉)的润滑剂种类,能制备稳定性和溶出行为良好,并且内容物的装量差异小、均匀度高的胶囊剂;具体表现为:
[0693]
所述胶囊剂中各杂质含量显著降低(0.005~0.07%,显著低于杂质限度标准≤0.1%),稳定性良好;溶出度为96~100%,高于限度标准≥85%,崩解速度快(30s),溶出行为显著;并且胶囊剂内容物的装量差异
±
1.8~2.8%,小于限度标准
±
5%,均匀度良好。
[0694]
②
其中,当采用润滑剂为硬脂酸镁时,制备得到的胶囊剂(例如,实施例1),制备的胶囊剂稳定性和溶出行为最优,装量差异最小、均匀度最好;
[0695]
所述胶囊剂各杂质含量低至0.005%,显著低于杂质限度标准≤0.1%,稳定性良好;溶出度高达100%,显著高于限度标准≥85%,崩解速度快(30s),溶出行为最显著;并且胶囊剂内容物的装量差异低至
±
1.8%,远小于限度标准
±
5%,均匀度优异。
[0696]
(十)、为了探究胶囊剂内容物形态的影响,在实施例1胶囊剂的内容物(囊芯)为颗粒的基础上,通过改变制备方法获得不同形态的填装内容物,改变为粉末(实施例11方法1)以及不同方法制备得到的颗粒(实施例11方法2)和微丸(实施例11方法3和4)进行比较。
[0697]
1、区别
[0698]
表48
[0699]
实施例胶囊剂内容物形态实施例1颗粒实施例11方法1粉末实施例11方法2颗粒实施例11方法3微丸实施例11方法4微丸实施例11方法5微片
[0700]
2、制剂对比
[0701]
表49
[0702][0703]
表50
[0704][0705]
表51
[0706][0707]
结论:
[0708]
1)杂质含量结果显示:
[0709]
新鲜制备条件下(0天):
[0710]
实施例1(颗粒)以及实施例11方法1(粉末)、实施例11方法2(颗粒)、实施例11方法3(微丸)和实施例11方法4(微丸)的pm4含量为0.006%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.006%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.10~0.12%,远低于限度标准(≤0.5%),稳定性良好。
[0711]
高温40℃条件下放置30天:
[0712]
实施例1(颗粒)以及实施例11方法1(粉末)、实施例11方法2(颗粒)、实施例11方法3(微丸)和实施例11方法4(微丸)的pm4含量为0.007~0.009%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.008%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.06%,远低于限度标准(≤0.1%);总杂含量为0.11~0.15%,远低于限度标准(≤0.5%),稳定性良好。
[0713]
高湿75%条件下放置30天:
[0714]
实施例1(颗粒)以及实施例11方法1(粉末)、实施例11方法2(颗粒)、实施例11方法3(微丸)和实施例11方法4(微丸)的pm4含量为0.006~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.007%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.05%,远低于限度标准(≤0.1%);总杂含量为0.11~0.13%,远低于限度标准(≤0.5%),稳定性良好。
[0715]
光照4500
±
500lx条件下放置30天:
[0716]
实施例1(颗粒)以及实施例11方法1(粉末)、实施例11方法2(颗粒)、实施例11方法3(微丸)和实施例11方法4(微丸)的pm4含量为0.007~0.009%,远低于限度标准(≤0.1%);杂质19含量为0.006~0.009%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.06%,远低于限度标准(≤0.1%);总杂含量为0.12~0.14%,远低于限度标准(≤0.5%),稳定性良好。
[0717]
上述结果表明,采用本发明的产品处方,不同的胶囊剂剂型均能得到内容物的稳定性良好的胶囊剂产品。
[0718]
2)溶出行为结果显示:
[0719]
实施例1(颗粒)得到的制剂在30s完全崩散,杯底无堆积,溶出度为100%,远高于溶出度限度(≥85%),溶出行为良好且显著。
[0720]
实施例11方法1(粉末)、实施例11方法2(颗粒)、实施例11方法3(微丸)和实施例11方法4(微丸)得到的制剂在30s完全崩散,杯底无堆积,溶出度为95~98%,较实施例11方法1(粉末)溶解度略有降低,但溶出度仍显著高于溶出度限度(≥85%),溶出行为良好。
[0721]
上述结果表明,采用本发明的产品处方,不同的胶囊剂剂型条件下,均能得到溶出行为良好的胶囊剂产品。
[0722]
3)胶囊填装结果显示:
[0723]
实施例1(颗粒)制备得到的胶囊内容物,颗粒流动性好,填充的重量更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0724]
实施例11方法1(粉末)、实施例11方法2(颗粒)、实施例11方法3(微丸)和实施例11方法4(微丸)制备得到的胶囊内容物,胶囊内容物的装量差异度控制在
±
2.2~3.5%,略有
升高,但仍控制在限度标准
±
5%范围内。
[0725]
上述结果表明,采用本发明的产品处方,不同的胶囊剂剂型均能得到内容物的装量差异优异且符合限度标准的胶囊剂产品。
[0726]
此外,实施例11方法5(微片)制备得到的胶囊剂,在新鲜制备条件和不同条件下放置情况下,杂质含量远低于限度标准,也显示出了良好的稳定性;并且溶出行为良好;胶囊内容物的装量差异小、均匀度高。实验数据与方法1~4结果保持一致,上述未展示。
[0727]
小结:
[0728]
采用本发明的产品处方,制备不同内容物形态(颗粒、粉末、微丸、微片)的胶囊剂,均能得到稳定性和溶出行为良好,装量差异小、均匀度高的胶囊剂;
[0729]
例如,实施例1(颗粒)、实施例11方法1(粉末)、实施例11方法2(颗粒)、实施例11方法3(微丸)、实施例11方法4(微丸)和实施例11方法5(微片)上述不同形态内容物的胶囊剂,均表现为稳定性和溶出行为良好,装量差异小、均匀度高;具体表现为:
[0730]
所述胶囊剂中各杂质含量显著降低(0.005~0.06%,显著低于杂质限度标准≤0.1%),稳定性良好;溶出度为95~100%,高于限度标准≥85%,崩解速度快(30s),溶出行为显著;并且胶囊剂内容物的装量差异
±
1.8~3.5%,小于限度标准
±
5%,均匀度良好。
[0731]
(十一)、为了探究胶囊剂中不同药物活性成分含量在制备胶囊剂过程中的普适性,在实施例1胶囊剂药物活性成分(以枸橼酸爱地那非计)的质量百分含量70wt%的基础上,通过改变药物活性成分的加入量,制备胶囊剂进行比较。
[0732]
1、区别
[0733]
表52
[0734]
实施例药物活性成分含量/wt%实施例170.0实施例12方法110.0实施例12方法240.0实施例12方法380.0
[0735]
2、制剂对比
[0736]
表53
[0737]
[0738][0739]
表54
[0740][0741]
表55
[0742][0743]
结论:
[0744]
1)杂质含量结果显示:
[0745]
新鲜制备条件下(0天):
[0746]
实施例1(药物活性成分含量=70.0wt%)以及实施例12方法1(药物活性成分含量=10.0wt%)、实施例12方法2(药物活性成分含量=40.0wt%)、实施例12方法3(药物活性成分含量=80.0wt%)的pm4含量为0.006~0.008%,远低于限度标准(≤0.1%);杂质19含量为0.005~0.009%,远低于限度标准(≤0.1%);其他最大单杂含量为0.04~0.08%,远低于限度标准(≤0.1%);总杂含量为0.10~0.15%,远低于限度标准(≤0.5%),稳定性良好。
[0747]
高温40℃条件下放置30天:
[0748]
实施例1(药物活性成分含量=70.0wt%)以及实施例12方法1(药物活性成分含量
=10.0wt%)、实施例12方法2(药物活性成分含量=40.0wt%)、实施例12方法3(药物活性成分含量=80.0wt%)的pm4含量为0.007~0.01%,远小于限度标准(≤0.1%);杂质19含量为0.006~0.01%,远小于限度标准(≤0.1%);其他最大单杂含量为0.05~0.07%,远低于限度标准(≤0.1%);总杂含量为0.11~0.19%,远低于限度标准(≤0.5%),稳定性良好。
[0749]
高湿75%条件下放置30天:
[0750]
实施例1(药物活性成分含量=70.0wt%)以及实施例12方法1(药物活性成分含量=10.0wt%)、实施例12方法2(药物活性成分含量=40.0wt%)、实施例12方法3(药物活性成分含量=80.0wt%)的pm4含量均为0.008~0.01%,远小于限度标准(≤0.1%);杂质19含量为0.007~0.01%,远小于限度标准(≤0.1%);其他最大单杂含量为0.05~0.06%,远低于限度标准(≤0.1%);总杂含量为0.12~0.19%,远低于限度标准(≤0.5%),稳定性良好。
[0751]
光照4500
±
500lx条件下放置30天:
[0752]
实施例1(药物活性成分含量=70.0wt%)以及实施例12方法1(药物活性成分含量=10.0wt%)、实施例12方法2(药物活性成分含量=40.0wt%)、实施例12方法3(药物活性成分含量=80.0wt%)的pm4含量为0.008~0.01%,远低于限度标准(≤0.1%);杂质19含量为0.008~0.02%,远低于限度标准(≤0.1%);其他最大单杂含量为0.05~0.08%,远低于限度标准(≤0.1%);总杂含量为0.13~0.19%,远低于限度标准(≤0.5%),稳定性良好。
[0753]
上述结果表明,采用本发明的产品处方,不同的药物活性成分用量均能得到内容物的稳定性良好的胶囊剂产品。
[0754]
2)溶出行为结果显示:
[0755]
实施例1(药物活性成分含量=70.0wt%)得到的制剂在30s完全崩散,杯底无堆积,溶出度为100%,远高于溶出度限度(≥85%),溶出行为良好且显著。
[0756]
实施例12方法1(药物活性成分含量=10.0wt%)、实施例12方法2(药物活性成分含量=40.0wt%)、实施例12方法3(药物活性成分含量=80.0wt%)得到的制剂在30s完全崩散,杯底少量堆积,溶出度为89~97%,较实施例1(药物活性成分含量=70.0wt%)溶出度略有降低,但溶出度仍显著高于溶出度限度(≥85%),溶出行为良好。
[0757]
上述结果表明,采用本发明的产品处方,不同的药物活性成分用量均能得到内容物的溶出行为良好的胶囊剂产品。
[0758]
3)胶囊填装结果显示:
[0759]
实施例1(药物活性成分含量=70.0wt%)制备得到的胶囊内容物,颗粒流动性好,填充的重量更均匀,胶囊内容物的装量差异小,仅为
±
1.8%,显著小于限度标准
±
5%。
[0760]
实施例12方法1(药物活性成分含量=10.0wt%)、实施例12方法2(药物活性成分含量=40.0wt%)、实施例12方法3(药物活性成分含量=80.0wt%)制备得到的胶囊内容物,胶囊内容物的装量差异度控制在
±
2.9~3.8%,略有升高,但仍控制在限度标准
±
5%范围内。
[0761]
上述结果表明,采用本发明的产品处方,不同的药物活性成分用量均能得到内容物的装量差异优异且符合限度标准的胶囊剂产品。
[0762]
小结:
[0763]
采用本发明的产品处方,制备不同药物活性成分含量(10~80wt%)的胶囊剂,均能得到稳定性和溶出行为良好,装量差异小、均匀度高的胶囊剂;
[0764]
例如,实施例1(药物活性成分含量=70.0wt%)、实施例12方法1(药物活性成分含量=10.0wt%)、实施例12方法2(药物活性成分含量=40.0wt%)、实施例12方法3(药物活性成分含量=80.0wt%)上述不同药物活性成分含量的胶囊剂,均表现为稳定性和溶出行为良好,装量差异小、均匀度高;具体表现为:
[0765]
所述胶囊剂中各杂质含量显著降低(0.005~0.08%,显著低于杂质限度标准≤0.1%),稳定性良好;溶出度为89~100%,高于限度标准≥85%,崩解速度快(30s),溶出行为显著;并且胶囊剂内容物的装量差异
±
1.8~3.8%,小于限度标准
±
5%,均匀度良好。
[0766]
实验例4生物利用度
[0767]
将本发明效果最好的代表性实施例1、不加酸性辅料的效果较差的实施例2以及不同胶囊剂内容物形态的实施例11方法1、方法2和方法3与市售枸橼酸爱地那非片剂(爱力士-30mg
×
2片)进行生物等效性考察,结果显示:本发明加入酸性辅料的枸橼酸爱地那非胶囊剂均具有生物利用度高的特点。具体如下:
[0768]
将实施例1、实施例2、实施例11的方法1和方法2的枸橼酸爱地那非胶囊与市售枸橼酸爱地那非片剂(爱力士-30mg
×
2片)在32例健康志愿者进行(空腹)双周期交叉试验设计,进行生物等效性研究。相关数据如下:
[0769]
表56
[0770][0771]
备注:
[0772]
t
max
:给药后出现血药达峰浓度的时间;
[0773]cmax
:给药后出现的血药达峰浓度;
[0774]
auc
last
:从零到无穷大时间内血药浓度-时间曲线下面积;
[0775]
auc
inf_obs
:从零到最后可测浓度血药浓度-时间曲线下面积;
[0776]
生物利用度/%=auc
last(实施例)
÷
auc
last(爱力士)
×
100%。
[0777]
结论:
[0778]
1)本发明不加酸性辅料的代表性实施例1(效果最好)和实施例2不加酸性辅料(效果差)与市售枸橼酸爱地那非片剂等效性对比。
[0779]
与爱力士-30mg
×
2片相比,实施例1的相对生物利用度显著提高了28.3%,而实施
例2的相对生物利用度显著降低,降低了13.1%。
[0780]
加入酸性辅料-酒石酸制备的枸橼酸爱地那非胶囊剂(实施例1),与爱力士-30mg
×
2片相比,体内均快速被吸收,1小时左右快速吸收达到临床所需的治疗窗的血药浓度,c
max
为299.18
±
35.5ng/ml,给药后出现的血药达峰浓度显著提高,t
max
为1.05
±
0.27h,血药达峰浓度的时间无显著性差异;auc
last
为1762.12
±
58.7ng
·
h/ml,与爱力士-30mg
×
2片的相对生物利用度为128.3%,说明本实施例1的枸橼酸爱地那非胶囊生物利用度相比普通片剂显著提高。
[0781]
实施例2不加酸性辅料制备的枸橼酸爱地那非胶囊(实施例2)与爱力士-30mg
×
2片相比,t
max
为1.22
±
0.52h,血药达峰浓度的时间无显著性差异,c
max
为235.23
±
26.5ng/ml,auc
last
为1193.32
±
29.5ng
·
h/ml,与爱力士-30mg
×
2片生物等效,相对生物利用度为86.9%,在生物等效性的80~125%区间范围内。
[0782]
2)本发明不同制剂形态的实施例11方法1(粉末)、方法2(颗粒)和方法3(微丸)与市售枸橼酸爱地那非片剂等效性对比。
[0783]
与爱力士-30mg
×
2片相比,实施例11方法1(粉末)、方法2(颗粒)和方法3(微丸)的相对生物利用度均呈现升高趋势,分别提高14.2%、15.1%和18.5%,提高效果显著。
[0784]
方法1胶囊内容物采用粉末直接灌装的工艺,方法2胶囊内容物采用干法制粒颗粒灌装、方法3胶囊内容物采用微丸灌装胶囊所得制剂1小时内快速吸收达到临床所需的治疗窗的血药浓度;c
max
分别为:274.48
±
31.6ng/ml、281.48
±
36.2ng/ml、276.28
±
23.5ng/ml。auc
last
分别为1568.35
±
43.7ng
·
h/ml、1580.35
±
36.2ng
·
h/ml、1627.42
±
32.5ng
·
h/ml;与爱力士-30mg
×
2片相比相对生物利用度为分别为114.2%、115.1%、118.5%,说明本实施例11的加酸性辅料的不同制剂形式的枸橼酸爱地那非胶囊生物利用度均显著提高。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1