用于治疗癌症和/或感染性疾病的TLR7和TLR8激动剂
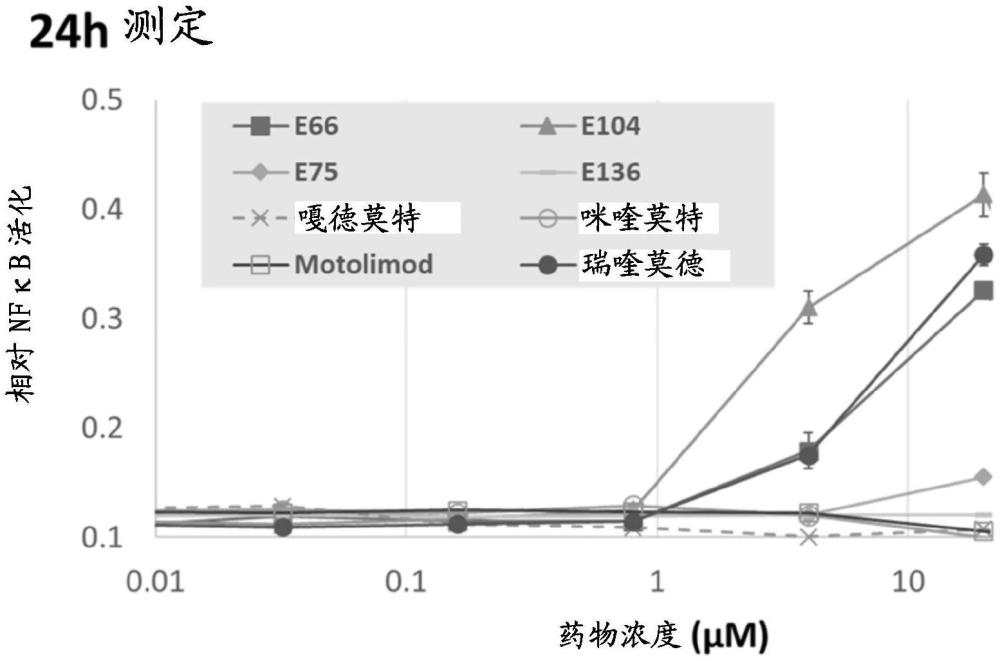
发明领域本发明涉及toll-样受体(tlr)的激动剂和toll-样受体(tlr)的激动剂的靶向递送。具体地,本发明的化合物是tlr 7和/或tlr8激动剂和抗体-药物-缀合物(adc),后者允许将tlr 7和/或tlr8激动剂递送和释放到期望的组织中,从而导致免疫活化。因此,本发明的化合物可用作用于治疗各种癌症和感染性疾病的治疗剂。发明背景toll-样受体(tlr)通过识别病原体相关分子模式(pamp)来控制先天性免疫系统应答。tlr7和tlr8属于已知的人tlr胞内体受体,并且能够诱导先天性免疫系统应答并且可以使用激动剂活化。tlr7和tlr8是结合来自内吞的细菌和病毒的单链rna并被其活化的同源受体。活化启动下游炎症应答,随后形成一种复合物,其启动信号传递级联并最终活化转录因子诸如活化的b细胞的核因子κ-轻链-增强子(nf-κb)和干扰素调节因子7(irf7)。这些然后刺激炎症性细胞因子和i型干扰素产生,它们对许多炎症过程很重要。由于tlr7和tlr8细胞因子诱导特性的差异以及免疫细胞类型之间的受体表达变异性,tlr7或tlr8的活化会导致独特的免疫应答。同样,由tlr7/8活化引起的细胞因子分泌有助于抗原特异性t和b细胞的活化,这有助于启动适应性免疫应答。tlr与许多免疫和炎性病症相关,并且因此,调节tlr活性的能力是治疗那些病症的潜在途径。tlr激动剂是经常用作疫苗佐剂的免疫刺激剂(参见,例如,mcgowan,d.,currenttopics in medicinal chemistry 19:2228-2238(2019))。这些激动剂活化适应性免疫系统,从而产生更稳健的抗病毒作用。tlr激动剂也被探索作为一种“揭露”免疫抑制性肿瘤环境的方法,希望免疫系统将癌症组织识别为“外来”并从而启动免疫系统的稳健抗肿瘤应答。tlr激动剂开发伴随着与炎症相关的副作用,商购可得的tlr7和tlr 8激动剂也是如此(参见kieffer等人,expert opinion on therapeutic patents 30(11):825-845(2020))。一种商购可得的tlr7激动剂是咪喹莫特。咪喹莫特已经被批准用于局部施用以治疗生殖器疣(抗病毒作用)、光化性角化病和非黑素瘤皮肤癌诸如基底细胞癌(抗肿瘤作用)。但是,由于全身给药的安全问题,咪喹莫特应用仅限于局部施用。如果全身递送的话,tlr激动剂诸如咪喹莫特会导致全身免疫刺激,从而导致细胞因子风暴型事件引起的急性毒性。咪喹莫特的两种imiquidazoline衍生物—瑞喹莫德(一种混合的tlr7/8激动剂,也被称作r848)和motolimod(一种tlr8激动剂,也被称作vtx-2337)—已经在多种临床前模型(包括癌症的免疫疗法模型)中显示出有前途的免疫刺激活性(参见,例如,prins等人,j.immunol.176:157-64(2006)和bialojan等人,eur.j.immunol.49:2083-2094(2019))。但是,这些激动剂尚未被监管机构批准用于治疗癌症患者。类似地,某些第三代tlr7/tlr8激动剂已经进入用于治疗病毒感染或癌症的临床开发,包括pf-4878691、bdc-1001、lhc165、nktr-262、tq-a3334、ro7119929、dsp-0509、bnt411和njh395(参见,例如,hanten等人,bmcimmunol.9:39(2008);weigel等人,am.j.hematol.87:953-956(2012);dudek等人,clin.cancer res.13:7119-7125(2007);fidock等人,clin.pharmacol.ther.89:821-829(2011);inglefield等人,j.interferon cytokine res.28:253-263(2008);astry等人,j.clin.pharmacol.48:755-762(2008);dummer等人,clin.cancer res.14:856-864(2008);harrison等人,j.clin.pharmacol.47:962-969(2007);bryden等人,sci.transl.med.12:eaax2421(2020);cromarty等人,front immunol.10:1705(2019);和larue等人,nat.rev.urol.10:537-545(2013))。但是,这些分子都未被监管部门批准用于人类。瑞喹莫德在治疗丙型肝炎病毒中的临床研究并不成功(pockros等人,j.hepatol.47(2):174-182(2007))。类似地,发现tlr7激动剂gsk-2245035在具有轻度过敏性哮喘的患者中缺乏改变过敏原诱导哮喘应答的效力(tsitoura等人,clin.pharmacol.ther.98(4):369-380(2015))。对tlr7激动剂pf-4878691的临床研究发现在治疗丙型肝炎病毒中具有低治疗指数(fidock等人,clin.pharmacol.ther.89(6):821-829(2011)),并且其它研究发现,tlr7激动剂gs-9620在hbv感染的原代人肝细胞中没有表现出抗病毒活性(tsai等人,j.virol.91(8):e02166-e16(2017)和bam等人,antimicrob.agents chemother.61(1):e01369-e16(2016))。寻找tlr7和tlr8激动剂的安全递送并同时维持治疗效果的方法是一个持续存在的问题。因此,有大量正在进行的研究致力于开发tlr7和tlr8激动剂的改进的递送平台,其实现稳健的局部递送而无需全身暴露。瑞喹莫德、motolimod以及其它tlr7和tlr8激动剂作为用于癌症患者的免疫刺激剂的开发面临着困难并且似乎陷入了僵局,至少部分地由于在最近临床试验中获得的令人失望的结果(frega等人,oncoimmunology 9:1-10(2020))。在该领域中的一个主要挑战是开发具有足够安全裕度的有效分子,因为tlr激动剂活化先天性免疫系统以增强人体的炎症应答(kieffer等人,expert opinion on therapeuticpatents30(11):825-845(2020);patel等人,future virol.9(9):811-829(2014);和tisoncik等人,microbiol.mol.biol.rev.76(1):16-32(2012))。因此,tlr激动剂在免疫肿瘤学中的应用是一个令人非常感兴趣的领域,但仍然非常需要改进的tlr7和tlr8激动剂。本公开内容旨在克服本领域的这些和其它缺陷。发明概述简而言之,本发明满足了对可以以局部方式递送、降低毒性并增强效力的靶向tlr激动剂的需要。在第一方面,本发明提供了式(i)或(ii)的化合物其中:r1选自c1-c10烷基、c1-c10氧杂烷基和c1-c10氮杂烷基;r2和r3各自独立地选自氢、c1-c5烷基和c1-c5烷氧基;n是1或2;y选自任选地被取代的芳基和任选地被取代的杂芳基;z1选自-nrz-、-o-、-nrzc(o)-、-nrzc(o)-o-和-nrzso2-;z2不存在,或选自(c1-c8)烃-nh-和5-8元含氮杂环,其中所述杂环的氮连接至x2;z选自-nrz-、-nrzc(o)-和-o-;rz选自氢、c1-c8烃、c1-c8氧杂烷基、c1-c8氮杂烷基、杂芳基和5-8元杂环;x1选自-rz、-c(o)-rz、-c(o)-o-rz、-c(o)-n-(rz)2、-(ch2)knrzc(o)-(c1-c6)烷基、-(ch2)knrzc(o)-o-(c1-c4)烷基和-so2-rz;k是1至8的整数;x2包含可切割的或不可切割的连接基;且ab包含抗体或抗体片段。在第二方面,本发明提供了式(iii)的化合物其中:r1选自c1-c10烷基、c1-c10氧杂烷基和c1-c10氮杂烷基;r2和r3各自独立地选自氢、c1-c5烷基和c1-c5烷氧基;n是1或2;y选自任选地被取代的芳基和任选地被取代的杂芳基;za选自-nrz-、-nrzc(o)-、-nrzc(o)-o-、-nrzc(o)-(ch2)k-nh-、-nrzc(o)-(ch2)k-o-、-nrzc(o)-o-(ch2)k-o-、-nrzc(o)-(ch2)k-n(ch3)-、-nrzc(o)-o-(ch2)k-nh-、-nrzc(o)-(ch2)k-nh-c(o)-o-和-nrzso2-;k是1至8的整数;rz选自氢、c1-c8烃、c1-c8氧杂烷基、c1-c8氮杂烷基、杂芳基和5-8元杂环;且xa选自氢、c1-c10烷基和-c(o)ch3。在第三方面,本发明提供了一种药物组合物,其包含本文描述的化合物和药学上可接受的载体、稀释剂或赋形剂。在第四方面,本发明提供了一种用于在受试者中刺激免疫应答的方法。所述方法包括在有效刺激免疫应答的条件下施用治疗有效量的本文描述的化合物。在第五方面,本发明提供了一种用于在受试者中诱导抗肿瘤免疫应答的方法。所述方法包括在有效诱导抗肿瘤免疫应答的条件下施用治疗有效量的本文描述的化合物。在第六方面,本发明提供了一种用于在受试者中治疗肿瘤或异常细胞增殖的方法。所述方法包括在有效治疗肿瘤或异常细胞增殖的条件下施用治疗有效量的本文描述的化合物。在第七方面,本发明提供了一种用于在受试者中治疗感染性疾病的方法。所述方法包括在有效治疗感染性疾病的条件下施用治疗有效量的本文描述的化合物。从以下结合附图对本发明各个方面的详细描述,本发明的这些和其它目的、特征和优点将变得显而易见。附图简述图1a和图1b显示了用本文公开的化合物处理24h(图1a)或72h(图1b)以后ramosblue细胞中nfκb的活化。图2证实了在mtlr7-hek293报道细胞系中的nfκb途径的活化。图3表明,抗-her2靶向的adc在培养基转移研究中活化tlr7报道系。图4表明,抗-her2 adc在培养基转移测定中活化mtl7。裸抗体的添加会抑制活性。图5证实,本文公开的adc在培养基转移测定中在1μg/ml以下显示出活性。图6显示了连接至e104和瑞喹莫德的替代连接基的评价。图7a和图7b证实了本文公开的化合物诱导的来自巨噬细胞(图7a)和单核细胞(图7b)的tnfα释放(通过elisa测量的)。图8显示了本文公开的三种adc的稳定性研究的结果,证实了在人血清和小鼠血清中温育期间有效负载的有限释放。图9显示了本文公开的adc在人和小鼠血清中的稳定性。图10证实了在小鼠中的异种移植物研究的结果,显示了本文公开的adc对肿瘤大小的影响。图11显示了本文公开的adc在小鼠异种移植物研究中对体重的影响。图12显示了本文公开的adc在人和小鼠血清中的稳定性。图13显示了本文公开的adc在人和小鼠血清中的稳定性。图14显示了本文公开的adc在人和小鼠血清中的稳定性。图15显示了本文公开的adc在人和小鼠血清中的稳定性。图16显示了本发明的抗-trop2和抗-gcc adc能够模拟在表达抗原的非小细胞肺癌组织附近的巨噬细胞的证据。图17显示了本发明的抗-trop2和抗-gcc adc能够模拟在表达抗原的胰腺癌组织附近的巨噬细胞的证据。发明详述几种高效的tlr激动剂已经连接至抗体导向的肿瘤细胞。抗体-药物-缀合物(adc)内化到肿瘤组织中,释放药物。该药物渗透到附近的组织,从而导致免疫活化。tlr激动剂的局部释放会诱导针对肿瘤的适应性免疫应答,从而驱动抗肿瘤作用。通过将tlr激动剂连接至与病原体结合的抗体,可以驱动抗病原体作用。然后,经调理的病原体将被树突细胞(抗原呈递细胞)吸收,在那里将释放tlr激动剂,从而导致增强的适应性免疫应答。在某些实施方案中,所述化合物是式(i)的化合物:在某些实施方案中,所述化合物是式(ii)的化合物:在某些实施方案中,所述化合物是式(iii)的化合物:在某些实施方案中,r1是c1-c10烷基。在其它实施方案中,r1是正丁基。在某些实施方案中,r1是c1-c10氧杂烷基。在其它实施方案中,r1是-ch2oh。在再其它的实施方案中,r1是-ch2ch2oh或r1是-ch2ch2ch2oh。在某些实施方案中,r1是-ch2och2ch3。在其它实施方案中,r1是-ch2och2ch2ch3或r1是-ch2ch2och2ch3或r1是-ch2ch2och3。在再其它的实施方案中,r1是c1-c10氮杂烷基。在某些实施方案中,r1是-ch2nhch2ch3。在其它实施方案中,r1是-ch2nhch2ch2ch3或r1是-ch2ch2nhch2ch3或r1是-ch2ch2nhch3。在某些实施方案中,n是1。在某些实施方案中,n是2。在某些实施方案中,y是未被取代的或被取代的芳基。在其它实施方案中,y是未被取代的或被取代的苯基。在某些实施方案中,y是未被取代的或被取代的杂芳基。在其它实施方案中,y是未被取代的或被取代的吡啶基。在某些实施方案中,y被卤素、c1-c4烷基、c1-c4烷氧基、c1-c4卤代烷基和/或c1-c4卤代烷氧基中的一种或多种取代。在其它实施方案中,y被氯、氟、甲基、乙基、丙基和/或甲氧基中的一种或多种取代。在某些实施方案中,z1是-nrz-。在其它实施方案中,z1是-o-。在某些实施方案中,z1是-nrzc(o)-。在某些实施方案中,z1是-nrzc(o)-o-。在再其它的实施方案中,z1是-nrzso2-。在某些实施方案中,z1是-nrz-或-o-。在某些实施方案中,z2不存在。在其它实施方案中,z2是-(c1-c8)烃-nh-。在再其它的实施方案中,z2是-(c1-c8)烷基-nh-。在其它实施方案中,z2是-苄基-nh-。在又其它实施方案中,z2是-苯基-nh-。在某些实施方案中,z2是5-8元含氮杂环,其中所述杂环的氮连接至x2。在某些实施方案中,z是-o-。在其它实施方案中,z是-nrz-。在再其它的实施方案中,z是-nrzc(o)-。在某些实施方案中,za是-nrz-。在某些实施方案中,za是-nrzc(o)-。在某些实施方案中,za是-nrzc(o)-o-。在其它实施方案中,za是-nrzc(o)-(ch2)k-nh-。在某些实施方案中,za是-nrzc(o)-(ch2)k-o-。在某些实施方案中,za是-nrzc(o)-o-(ch2)k-o-。在其它实施方案中,za是-nrzc(o)-(ch2)k-n(ch3)-。在再其它的实施方案中,za是-nrzc(o)-o-(ch2)k-nh-。在某些实施方案中,za是-nrzc(o)-(ch2)k-nh-c(o)-o-。在某些实施方案中,za是-nrzso2-。在某些实施方案中,x1是rz。在某些实施方案中,x1是氢。在某些实施方案中,x1是甲基。在其它实施方案中,x1是c(o)-rz。在再其它的实施方案中,x1是c(o)-o-rz。在某些实施方案中,x1是c(o)-n-(rz)2。在其它实施方案中,x1是so2-rz。在其它实施方案中,x1是-(ch2)knrzc(o)-(c1-c6)烷基。在又其它实施方案中,x1是-(ch2)knrzc(o)-o-(c1-c4)烷基。在某些实施方案中,rz是氢。在其它实施方案中,rz是c1-c8烃。在再其它的实施方案中,rz是c1-c8烷基。在某些实施方案中,rz是甲基。在其它实施方案中,rz是c1-c8氧杂烷基。在再其它的实施方案中,rz是c1-c8氮杂烷基。在又其它实施方案中,rz是-c(nh2)苄基。在某些实施方案中,rz是杂芳基。在某些实施方案中,rz是5-8元杂环。独立地选择rz的每个实例。作为非限制性的示例性实施例,如果z是-nrz-且x1是-c(o)-rz,rz的两个实例可以是氢,或z可以是-nch2-且x1可以是-c(o)ch3,或z可以是-nh-且x1可以是-c(o)ch2ch3。在某些实施方案中,k是1至8的整数。在某些实施方案中,k是1至6的整数。在某些实施方案中,k是1至4的整数。在某些实施方案中,k是1至3的整数。在某些实施方案中,k是1至2的整数。在某些实施方案中,k是2至4的整数。在某些实施方案中,k是2至3的整数。在某些实施方案中,k是1。在某些实施方案中,k是2。在某些实施方案中,k是3。在某些实施方案中,k是4。在某些实施方案中,k是5。在某些实施方案中,k是6。在某些实施方案中,k是7。在某些实施方案中,k是8。在某些实施方案中,xa是氢。在某些实施方案中,xa是c1-c10烷基。在某些实施方案中,xa是c1-c4烷基。在某些实施方案中,xa是甲基。在某些实施方案中,xa是乙基。在某些实施方案中,xa是丙基。在某些实施方案中,xa是丁基。在某些实施方案中,xa是正丁基。在某些实施方案中,xa是叔丁基。在某些实施方案中,xa是-c(o)ch3。在某些实施方案中,当所述化合物具有式(i)、x1是氢、n是1、且y是苯基时,所述化合物具有式(ia)、(ib)、(ic)、(id)、(ie)、(if)、(ig)或(ih):在式(ia)、(ib)、(ic)、(id)、(ie)、(if)、(ig)或(ih)的这些实施方案中,代表与x2的连接点。在某些实施方案中,当所述化合物具有式(ii)、n是1、且y是苯基时,所述化合物具有式(iia)、(iib)、(iic)、(iid)、(iie)、(iif)、(iig)或(iih):其中代表与x2的连接点。在某些实施方案中,r2是氢。在其它实施方案中,r2是c1-c5烷基。在某些实施方案中,r2选自甲基、乙基、丙基或丁基。在再其它的实施方案中,r2是c1-c5烷氧基。在某些实施方案中,r2选自甲氧基、乙氧基、丙氧基或丁氧基。在某些实施方案中,r3是氢。在其它实施方案中,r3是c1-c5烷基。在某些实施方案中,r3选自甲基、乙基、丙基或丁基。在再其它的实施方案中,r3是c1-c5烷氧基。在某些实施方案中,r2选自甲氧基、乙氧基、丙氧基或丁氧基。为了非常清楚起见,独立地选择r2和r3。作为非限制性例子,r2和r3可以是氢,r2和r3可以是c1-c5烷基,或r2和r3中的一个可以是c1-c5烷氧基且r2和r3中的另一个可以是氢。在一个实施方案中,x2是l1-l2-(l3)p-(l4)q-(l5)r。在一个实施方案中,式(i)或式(ii)的化合物的x2是可切割的或不可切割的连接基(在本文中被称作“连接基”或“l”,如在本文中进一步描述的)。所述连接基可以是可切割的,由以下连接基组成:化学不稳定的连接基,包括酸可切割的连接基和可还原的连接基,或酶可切割的连接基,诸如本领域众所周知的基于肽的连接基或葡糖苷酸连接基。在一个实施方案中,所述连接基可通过胞内酶(例如,组织蛋白酶-b或天冬酰胺肽链内切酶(legumain))切割。在adc中,所述连接基用于将有效负载连接至所述抗体。在一个实施方案中,引入所述连接基单元的第二部分,其具有第二反应位点,例如可与在抗体单元(例如,抗体)上存在的亲核基团反应的亲电子基团。在抗体上有用的亲核基团包括、但不限于巯基、羟基和氨基基团。抗体的亲核基团的杂原子可以与连接基单元上的亲电子基团反应并与连接基单元形成共价键。有用的亲电子基团包括、但不限于马来酰亚胺、卤代乙酰胺和活化的酯基团。所述亲电子基团可以为抗体连接提供方便的位点。在另一个实施方案中,连接基单元具有反应位点,所述反应位点具有可与在抗体上存在的亲电子基团反应的亲核基团。在抗体上有用的亲电子基团包括、但不限于醛和酮羰基基团。连接基单元的亲核基团的杂原子可以与抗体上的亲电子基团反应并与抗体形成共价键。在连接基单元上有用的亲核基团包括、但不限于酰肼、肟、氨基、肼、缩氨基硫脲、羧酸肼和芳基酰肼。在抗体上的亲电子基团可以提供用于连接连接基单元的方便位点。在另一个实施方案中,连接基单元具有可以通过酶促反应连接至抗体的官能团。这方面的一个特别有用的例子是含胺连接基与谷氨酰胺的转酰胺基作用,这是由细菌转谷氨酰胺酶促进的反应。该反应可以用于将有效负载连接至内源性谷氨酰胺残基,如benjamin等人(mol.pharmaceutics 2019,16,6,2795-2807)所述,或可以用于将有效负载连接至专门工程改造的谷氨酰胺标签,如strop等人(chemistry and biology 2013,20,2,161-167)所述,它们二者特此通过引用整体并入。氨基官能团也是连接基单元的有用反应位点,因为它们可以与羧酸或化合物的活化酯反应以形成酰胺键。在一个实施方案中,本公开内容的基于肽的化合物可以通过在两个或更多个氨基酸和/或肽片段之间形成肽键来制备。这样的肽键可以例如根据液相合成方法(参见,例如,schroder和lubke,the peptides,第1版,第76-136页(academic press1966,其特此通过引用整体并入)来制备,这在肽化学领域中是众所周知的。在本公开内容的上下文中,特别但不限于连接基组分,措辞“选自……中的一种或多种”或“……中的一种或多种”指示,可以相同或不同的多个组分是或可以是按顺序排列。因此,例如,l2可以是任何单独或组合列出的组分。根据本公开内容,将式(i)或式(ii)的连接基定义为x2,并且在某些实施方案中,x2是l1-l2-(l3)p-(l4)q-(l5)r。根据本公开内容的一个实施方案,l1是缀合部分。本文描述的缀合部分包括将本文描述的l2连接至半胱氨酸、赖氨酸或谷氨酰胺残基的部分。在某些实施方案中,所述谷氨酰胺是谷氨酰胺295。l1的例子包括马来酰亚胺、溴乙酰胺、胺、nhs-酯等。在一个实施方案中,如本文描述的l2是间隔基单元,其选自分支的或未分支的c1-c12烷基、选自peg1至peg12的peg、peg可以例如是peg1、peg2、peg3、peg4、peg5、peg6、peg7、peg8、peg9、peg10、peg11、peg12或它们的任何组合。在一个实施方案中,如本文描述的l3涉及1至6个氨基酸的肽。例如,所述肽可以是1个氨基酸、2个氨基酸、3个氨基酸、4个氨基酸、5个氨基酸或6个氨基酸。氨基酸可以选自天然的氨基酸和非天然的α-氨基酸。在一个实施方案中,l4是自降解的间隔基。在一个实施方案中,l5是如本文描述的羰基。在一个实施方案中,式(i)和式(ii)的化合物的p、q和r各自独立地选自0和1。在一个实施方案中,式(i)或式(ii)的化合物可以包括选自以下的l1:在另一个实施方案中,式(i)或式(ii)的化合物可以包括选自的l2。在又另一个实施方案中,式(i)或式(ii)的化合物可以包括的l3,其中r是氨基酸侧链。在又另一个实施方案中,式(i)或式(ii)的化合物可以包括选自以下的l4:在又另一个实施方案中,式(i)或式(ii)的化合物可以包括的l5;且p、q和r各自独立地是0或1,其中当q是0时,则r是0,或当p是1时,则r是1。在某些实施方案中,l3可以包括、但不限于valcit、glyvalcit、valarg、phelys、alaala、glyglyphegly、alaalaala、alaasn、asnasn、asnala、valcitglypro、asnglypro、asnasnglypro、asn、glyasn、asnala、procitala、proasnleu、proasnala、propheala、prophegly、procitleu、proasnpro、proasnser和proasngly。在一个实施方案中,式(i)或式(ii)的化合物可以包括选自以下的l1:为的l2;为valcit、glyvalcit、asnasn、asn或alaala的l3;选自以下的l4:和/或为的l5;且p、q和r各自是0,或p、q和r各自是1。在本文描述的化合物中,在一个实施方案中,x2可以通过ab的半胱氨酸残基、ab的赖氨酸残基或ab的谷氨酰胺残基连接至ab。在某些实施方案中,所述谷氨酰胺是谷氨酰胺295。在某些实施方案中,x2通过ab的谷氨酰胺残基连接至ab,其中所述谷氨酰胺是谷氨酰胺295,且l1是除非明确说明,否则式(i)或式(ii)或式(iii)的化合物包括式(ia)、(ib)、(ic)、(id)、(ie)、(if)、(ig)、(ih)或(iia)、(iib)、(iic)、(iid)、(iie)、(iif)、(iig)、(iih)或(iii)的化合物。除非明确说明,否则本文描述的实施方案涉及式(i)或式(ii)或式(iii)中的任一个,或式(ia)、(ib)、(ic)、(id)、(ie)、(if)、(ig)、(ih)、(iia)、(iib)、(iic)、(iid)、(iie)、(iif)、(iig)、(iih)或(iii)中的任一个。在一个方面,本发明提供了一种药物组合物,其包含本文描述的化合物和药学上可接受的载体、稀释剂或赋形剂。在一个实施方案中,所述药物组合物进一步包含治疗有效量的化学治疗剂。在一个方面,本发明提供了一种在受试者中刺激免疫应答的方法。所述方法包括在有效刺激免疫应答的条件下施用治疗有效量的本文描述的化合物。在某些实施方案中,对具有癌症的受试者执行所述方法。在其它实施方案中,所述癌症是膀胱癌、乳癌、宫颈癌、结肠癌、子宫内膜癌、肾癌、肺癌、食管癌、卵巢癌、前列腺癌、胰腺癌、皮肤癌、胃癌、睾丸癌、胆管癌、结肠直肠癌、子宫内膜癌、头/颈癌、甲状腺髓样癌(medullary thyroid cancer)、肾癌、眼癌、神经母细胞瘤、蕈样肉芽肿病、神经胶质肿瘤、其它脑肿瘤、脊髓肿瘤、肝癌、白血病、淋巴瘤或它们的任何组合。在某些实施方案中,将存在于液体药物组合物中的免疫治疗化合物施用到肿瘤中(例如,肿瘤内(it)施用)并诱导针对肿瘤抗原的先天性免疫应答和细胞介导的免疫应答(例如,收缩或稳定肿瘤)。所述包含肽的缀合物不一定是抗原或免疫原,而是降低tlr7和/或tlr8激动剂的溶解度的机制,从而产生被保留在施用部位处(诸如在肿瘤内或在肿瘤微环境中)的贮库。缀合的tlr7和/或tlr8激动剂可以刺激免疫抑制细胞并且可以诱导针对在肿瘤中存在的抗原的免疫应答。此外,免疫抑制细胞的动员不仅可以诱导针对施用部位处的肿瘤的免疫应答,而且还可以诱导针对周围的、附近的和/或远处的肿瘤的免疫应答。在一个实施方案中,公开了在受试者中刺激抗肿瘤免疫应答的方法,其中所述方法包含向所述受试者中局部地在肿瘤内或在肿瘤旁施用液体形式的药物组合物,其中所述抗肿瘤免疫应答在远离所述药物组合物的施用部位的部位是有效的。在第四方面,本发明提供了一种用于在受试者中诱导抗肿瘤免疫应答的方法。所述方法包括在有效诱导抗肿瘤免疫应答的条件下施用治疗有效量的本文描述的化合物。在某些实施方案中,对具有肿瘤的选定受试者执行所述方法。在某些实施方案中,所述肿瘤是纤维肉瘤、粘液肉瘤、脂肉瘤、软骨肉瘤、成骨性肉瘤、脊索瘤、血管肉瘤、内皮肉瘤、淋巴管肉瘤、淋巴管内皮肉瘤、滑膜瘤、间皮瘤、尤因瘤、平滑肌肉瘤、横纹肌肉瘤、结肠癌、胰腺癌、乳癌、卵巢癌、前列腺癌、鳞状细胞癌、基底细胞癌、腺癌、汗腺癌、皮脂腺癌、乳头状癌、乳头状腺癌、囊腺癌、髓样癌、支气管原癌、肾细胞癌、肝细胞瘤、胆管癌、绒毛膜癌、精原细胞瘤、胚胎性癌、威尔曼瘤、宫颈癌、睾丸肿瘤、肺癌、小细胞肺癌、膀胱癌、上皮癌、神经胶质瘤、星形细胞瘤、髓母细胞瘤、颅咽管瘤、室管膜瘤、松果体瘤、血管母细胞瘤、听神经瘤、少突神经胶质瘤、脑膜瘤、黑素瘤、神经母细胞瘤或视网膜母细胞瘤。在第五方面,本发明提供了一种用于在受试者中治疗肿瘤或异常细胞增殖的方法。所述方法包括在有效治疗肿瘤或异常细胞增殖的条件下施用治疗有效量的本文描述的化合物。在某些实施方案中,所述肿瘤或异常细胞增殖是癌症。在某些实施方案中,所述癌症是膀胱癌、乳癌、宫颈癌、结肠癌、子宫内膜癌、肾癌、肺癌、食管癌、卵巢癌、前列腺癌、胰腺癌、皮肤癌、胃癌、睾丸癌、胆管癌、结肠直肠癌、子宫内膜癌、头颈癌、甲状腺髓样癌、肾癌、眼癌、神经母细胞瘤、蕈样肉芽肿病、神经胶质肿瘤、其它脑肿瘤、脊髓肿瘤、肝癌、白血病、淋巴瘤或它们的任何组合。在第六方面,本发明提供了一种用于在受试者中治疗感染性疾病的方法。所述方法包括在有效治疗感染性疾病的条件下施用治疗有效量的本文描述的化合物。在某些实施方案中,所述感染性疾病是病毒感染、细菌感染、真菌感染或它们的任何组合。在某些实施方案中,所述感染性疾病是病毒感染,且所述感染性疾病是冠状病毒(包括、但不限于严重急性呼吸综合征(sars)、sars-cov-2(covid-19)、中东呼吸综合征(mers)和普通感冒)、埃博拉、流感、肝炎、hib疾病、人免疫缺陷病毒(hiv)、人乳头瘤病毒(hpv)、脑膜炎球菌疾病、肺炎球菌疾病、麻疹、腮腺炎、诺如病毒、脊髓灰质炎、呼吸道合胞病毒(rsv)、轮状病毒、风疹病毒、带状疱疹、西尼罗河病毒、狂犬病病毒、肠道病毒、巨细胞病毒、疱疹病毒、水痘、黄热病、寨卡病毒或它们的任何组合。在某些实施方案中,所述感染性疾病是细菌感染,且所述感染性疾病选自链球菌疾病、葡萄球菌疾病、白喉、脑膜炎球菌疾病、破伤风、百日咳、肺炎球菌疾病、细菌食品中毒、性传播的感染、结核病、莱姆病、肉毒中毒或它们的任何组合。在某些实施方案中,所述感染性疾病是真菌感染,且所述感染性疾病是念珠菌病、组织胞浆菌病、皮肤癣菌病、足癣、曲霉菌病、隐球菌性脑膜炎、球孢子菌病或它们的任何组合。缩写和定义除非另外定义,否则在本文中使用的所有技术和科学术语具有与本公开内容所属领域的普通技术人员通常理解的相同的含义。有机化学家(即,本领域普通技术人员)所使用的缩写词的综合列表出现在journal of organic chemistry的每卷的第一期中。通常呈现在标题为“标准缩写列表(standard list of abbreviations)”的表中的列表通过引用并入本文。在本文引用的术语存在多个定义的情况下,除非另外说明,否则以在本部分中的定义为准。以下缩写和术语自始至终具有指出的含义:ac =乙酰基aq =水性的boc =叔丁基氧基羰基bu =丁基c- =环dcm =二氯甲烷=二氯甲烷=ch2cl2dma =二甲基乙酰胺dmf =n,n-二甲基甲酰胺eq.或equiv.= 当量et =乙基h =小时hatu =六氟磷酸盐氮杂苯并三唑四甲基脲鎓hobt =羟基苯并三唑mc =马来酰亚胺基己酰基mcpba =间氯过氧苯甲酸me =甲基min.= 分钟pab =4-氨基苄基pabc=对氨基苄基氨基甲酸酯pg =保护基团ph =苯基pnp =对硝基苯酚rt =室温sat’d或sat.= 饱和的seap=分泌的胚胎碱性磷酸酶std=标准差t-或tert=叔tfa =三氟乙酸thf =四氢呋喃tosyl =对甲苯磺酰基uplc=超高效液相色谱法本文中使用的术语“包含”和“包括”或其语法变体应被视为指定所述特征、整数、步骤或组分,但不排除一个或多个另外特征、整数、步骤、组分或其组的添加。该术语涵盖术语“由……组成”和“基本上由……组成”。短语“基本上由……组成”或其语法变体当在本文中使用时应被视为指定所述特征、整数、步骤或组分,但不排除一个或多个另外特征、整数、步骤、组分或其组的添加,但前提是,另外特征、整数、步骤、组分或其组不实质上改变要求保护的组合物或方法的基本和新颖特征。为了本公开内容的目的,术语“抗体”(或“ab”或“ab”)在本文中在最宽的含义上使用,并且具体涵盖完整的单克隆抗体、多克隆抗体、单特异性抗体、多特异性抗体(例如,双特异性抗体)、表现出所需生物活性的抗体片段、抗体的遗传工程改造形式及其组合。此外,虽然本公开内容的某些方面涉及抗体药物缀合物,但考虑到缀合物的抗体部分可以被这样的任何物质替代:该物质与给定的靶细胞群体相关联的受体、抗原或其它接受部分特异性地结合或反应性地结合或复合。例如,本公开内容的缀合物可以包括靶向分子,其与寻求治疗学上或以其它方式生物学上修饰的细胞群体的受体、抗原或其它接受部分结合、复合或反应。这样的分子的例子包括小分子量蛋白、多肽或肽、凝集素、糖蛋白、非肽、维生素、营养物运输分子(例如、转铁蛋白)或任何其它细胞结合分子或物质。在某些方面,所述抗体或其它这样的靶向分子用于将药物递送至所述抗体或其它靶向分子所相互作用的特定靶细胞群体。在一个实施方案中,“ab”包含抗体或抗体片段。虽然本文公开了抗体(即,“ab”)的某些具体例子,但如技术人员将理解的,可以成功使用的抗体不限于这些例子。与术语“免疫球蛋白”互换使用的术语“抗体”包括全长(即,天然存在的或通过正常免疫球蛋白基因片段重组过程形成的)免疫球蛋白分子(例如,igg抗体)及其免疫活性片段(即,包括全长免疫球蛋白分子的特异性结合部分),其同样在性质上可以是天然存在的或合成的。因此,术语“抗体片段”包括抗体的一部分,诸如f(ab′)2、f(ab)2、fab′、fab、fv、scfv等。无论结构如何,抗体片段与全长抗体所识别的相同抗原结合。制备和筛选抗体片段的方法是本领域众所周知的。天然存在的抗体通常具有两条相同的重链和两条相同的轻链,每条轻链通过链间二硫键与重链共价连接,并且多个二硫键进一步将两条重链彼此连接。各个链可以折叠成具有相似大小(110-125个氨基酸)和结构但不同功能的结构域。所述轻链可以包含一个可变结构域(vl)和/或一个恒定结构域(cl)。所述重链还可以包含一个可变结构域(vh)和/或(取决于抗体的类别或同种型)三个或四个恒定结构域(ch1、ch2、ch3和ch4)。所述可变区与靶抗原结合并相互作用。所述可变区包括互补决定区(cdr),其识别并结合特定抗原上的特异性结合位点。所述恒定区可以被免疫系统识别并与其相互作用(参见,例如,janeway等人,immunobiology,第5版,garland science(纽约2001),其特此通过引用整体并入)。抗体可以是任何类型或类别(例如,igg、ige、igm、igd和iga)或亚类(例如,igg1、igg2、igg3、igg4、igal和iga2)。在人类中,所述同种型是iga、igd、ige、igg和igm,其中iga和igg进一步细分为亚类或亚型(iga1-2和igg1-4)。抗体可以源自任何合适的物种。在某些实施方案中,所述抗体具有人或鼠起源。抗体可以是,例如,人的、人源化的或嵌合的。通常,所述可变结构域从一种抗体到下一种抗体显示出相当大的氨基酸序列变异性,特别是在抗原结合位点的位置处。在vl和vh中各发现了三个区域,称为高变区或互补性决定区(cdr),它们由被称为框架可变区的不太可变区域支持。抗体包括igg单克隆抗体以及抗体片段或经工程改造的形式。这些是例如fv片段或蛋白,其中示例性抗体的cdr和/或可变结构域被工程改造为单链抗原结合蛋白。抗体的由vl和vh结构域组成的部分被命名为fv(可变片段)并构成抗原结合位点。单链fv(scfv或sca)是在一条多肽链上含有vl结构域和vh结构域的抗体片段,其中一个结构域的n端和另一个结构域的c端通过柔性连接基连接。用于产生单链抗体的肽连接基通常是柔性肽,其经过选择以确保发生vl和vh结构域的适当三维折叠。所述连接基通常为10至50个氨基酸残基,并且在某些情况下更短,例如,约10至30个氨基酸残基、或12至30个氨基酸残基、或甚至15至25个氨基酸残基。这样的连接基肽的一个例子包括四个甘氨酸残基的重复,随后是一个丝氨酸残基。单链抗体缺乏衍生出它们的完整抗体的部分或全部恒定结构域。因此,他们可以克服与完整抗体的使用相关的一些问题。例如,单链抗体往往不存在重链恒定区和其它生物分子之间的某些不希望的相互作用。此外,单链抗体比完整抗体小得多,并且可以具有比完整抗体更大的渗透性,从而允许单链抗体更高效地定位并结合靶抗原结合位点。此外,与完整抗体相比,单链抗体的相对小尺寸使得它们不太可能在接受者中引发不希望的免疫应答。fab(片段,抗原结合)表示由vl、cl、vh和ch1结构域组成的抗体片段。在木瓜蛋白酶消化后产生的那些被简称为fab,并且不保留重链铰链区。在胃蛋白酶消化后,会产生各种保留重链铰链的fab。链间二硫键保持完整的那些片段被称作f(ab′)2,而当二硫键不保留时,产生单个fab'。f(ab′)2片段对抗原的亲和力比单价fab片段更高。fc(片段结晶)是包含配对的重链恒定结构域的抗体部分或片段的名称。例如,在igg抗体中,所述fc包含ch2和ch3结构域。iga或igm抗体的fc进一步包含ch4结构域。fc与fc受体结合、补体介导的细胞毒性和抗体依赖性的细胞的细胞毒性(adcc)的活化相关。对于抗体诸如iga和igm(它们是多个igg-样蛋白的复合物),复合物形成需要fc恒定结构域。最后,所述铰链区将抗体的fab和fc部分分开,从而提供fab相对于彼此以及相对于fc的移动性,以及包括用于两条重链共价连接的多个二硫键。抗体“特异性”表示抗体对抗原的特定表位的选择性识别。术语“表位”包括能够特异性结合免疫球蛋白或t细胞受体或以其它方式与分子相互作用的任何蛋白决定簇。表位决定簇通常由分子的化学活性表面基团(诸如氨基酸或碳水化合物或糖侧链)组成,并且通常具有特定的三维结构特征以及特定的电荷特征。表位可以是“线性的”或“构象的”。在线性表位中,蛋白和相互作用分子(诸如抗体)之间的所有相互作用点沿着蛋白的一级氨基酸序列线性发生。在构象表位中,所述相互作用点发生在蛋白上彼此分离的氨基酸残基之间,即通过蛋白的三级折叠并置的非连续氨基酸。由连续氨基酸形成的表位通常在暴露于变性溶剂时保留,而由三级折叠形成的表位通常在用变性溶剂处理时丢失。表位通常包括在独特空间构象中的至少3个、且更常见地至少5个或8-10个氨基酸。识别相同表位的抗体可以通过简单的免疫测定进行验证,显示出一种抗体阻断另一种抗体与靶抗原结合的能力。如本文描述的,短语“特异性地结合”和“特异性结合”表示抗体与预定抗原结合。有用的多克隆抗体是从经免疫的动物的血清衍生出的抗体分子的异质群体。有用的单克隆抗体是针对特定抗原决定簇(例如,癌细胞抗原、病毒抗原、微生物抗原、蛋白、肽、碳水化合物、化学物、核酸或其片段)的抗体的均匀群体。使用本领域已知的任何技术可以制备针对目标抗原的单克隆抗体(mab),所述技术通过培养的连续细胞系提供抗体分子的生产。本文中使用的术语“单克隆抗体”表示从基本上均匀的抗体群体得到的抗体,即,除了可能以微小量存在的可能的天然存在的突变以外,构成所述群体的各个抗体是相同的。单克隆抗体是高度特异性的,针对单一抗原位点。修饰词“单克隆的”指示所述抗体得自基本上同源的抗体群体的特征,并且不应解释为需要通过任何特定方法生产所述抗体。单克隆抗体可以是鼠的、人的、人源化的或嵌合的。人源化抗体是一种重组蛋白,其中抗体的cdr来自一个物种;例如,将啮齿动物、兔、狗、山羊、马或鸡抗体(或任何其它合适的动物抗体)转移进人重和轻可变结构域中。抗体分子的恒定结构域源自人抗体的恒定结构域。制备人源化抗体的方法是本领域众所周知的。嵌合抗体优选地具有基本上或排它地源自人抗体恒定区的恒定区以及基本上或排它地源自除人之外的哺乳动物的可变区序列的可变区。通过还用相应的人序列替换鼠(或其它非人哺乳动物)抗体的除高变区或互补性决定区(cdr)之外的可变区,可以使嵌合过程更有效。除cdr之外的可变区也被称作可变框架区(fr)。术语“单克隆抗体”具体包括“嵌合”抗体,其中重链和/或轻链的一部分与源自特定物种或属于特定抗体类别或亚类的抗体的相应序列相同或同源,而所述链的其余部分与源自另一个物种或属于另一个抗体类别或亚类的抗体的相应序列相同或同源,以及这样的抗体的片段,只要它们表现出期望的生物活性。有用的单克隆抗体包括、但不限于人单克隆抗体、人源化的单克隆抗体、抗体片段或嵌合的单克隆抗体。通过本领域已知的众多技术中的任一种,可以制备人单克隆抗体(例如,teng等人,“construction and testing of mouse--human heteromyelomas forhuman monoclonal antibody production,”proc.natl.acad.sci.usa 80:7308-12(1983);kozbor等人,“the production of monoclonal antibodies from humanlymphocytes,”immunology today 4:72-79(1983);和olsson等人,“human--humanmonoclonal antibody-producing hybridomas:technical aspects,”meth.enzymol.92:3-16(1982),它们都特此通过引用整体并入)。所述抗体也可以是双特异性抗体。用于制备双特异性抗体的方法是本领域已知的并且在本文中进行了讨论。如本文描述的“完整抗体”包括包含抗原结合可变区以及轻链恒定结构域(cl)和重链恒定结构域、cm、ch2、cm和ch4(视抗体类别而定)的抗体。所述恒定结构域可以是天然序列恒定结构域(例如,人天然序列恒定结构域)或其氨基酸序列变体。完整抗体可以具有一种或多种“效应物功能”,其表示可归因于抗体的fc区(例如,天然序列fc区或氨基酸序列变体fc区)的那些生物活性。抗体效应物功能的例子包括补体依赖性的细胞毒性、抗体依赖性的细胞介导的细胞毒性(adcc)和抗体依赖性的细胞介导的吞噬作用。参见pfizer inc.的wo 2014/068443,其特此通过引用整体并入。“抗体片段”包含完整抗体的一部分,优选地包含其抗原结合区或可变区。所述抗体可以是免疫特异性地结合靶细胞(例如,癌细胞抗原、病毒抗原或微生物抗原)的抗体的在功能上有活性的片段、衍生物或类似物,或结合肿瘤细胞或基质的其它抗体。在这点上,“在功能上有活性的”是指,所述片段、衍生物或类似物能够引发抗-抗-独特型抗体,所述抗体识别与衍生出所述片段、衍生物或类似物的抗体所识别的抗原相同的抗原。具体地,在一个示例性实施方案中,通过删除位于特异性地识别抗原的cdr序列的c端的框架和cdr序列,可以增强免疫球蛋白分子的独特型的抗原性。为了确定哪些cdr序列结合抗原,可以通过本领域已知的任何结合测定方法(例如,bia芯测定)将含有cdr序列的合成肽与抗原一起用在结合测定中(对于cdr序列的定位,参见,例如,kabat等人,sequencesof proteinsof immunologicalinterest,第五版,national institute of health(bethesda,md.1991);kabat e.,“origins of antibody complementarity and specificity--hyper可变regions andminigene hypothesis,”j.immunology 125(3):961-969)(1980),它们二者特此通过引用整体并入)。抗体片段的例子包括fab、fab'、f(ab')2和fv片段、双体、三体、四体、直链抗体、单链抗体分子、scfv、scfv-fc、由抗体片段形成的多特异性抗体片段、由fab表达文库产生的片段、与所述抗体具有相同特异性的任何其它分子、或免疫特异性地结合靶抗原(例如,癌细胞抗原、病毒抗原或微生物抗原)的任何上述分子的表位结合片段。术语“可变”在抗体的上下文中表示抗体的可变结构域的某些部分,其序列广泛不同并且用于每种特定抗体对其特定抗原的结合和特异性。这种变异性集中在轻链和重链可变结构域中被称作“高变区”的三个区段中。可变结构域的更高度保守的部分被称作框架区(fr)。天然重链和轻链的可变结构域各自包含由三个高变区连接的四个fr。本文中使用的短语“高变区”包括抗体的负责抗原结合的氨基酸残基。所述高变区通常包含来自“互补性决定区”或“cdr”的氨基酸残基(例如,在轻链可变结构域中的残基24-34(li)、50-56(l2)和89-97(l3)以及在重链可变结构域中的残基31-35(hl)、50-65(h2)和95-102(l3)(kabat等人,sequencesof proteinsof immunological interest,第五版,nationalinstitute of health(bethesda,md.1991),其特此通过引用整体并入);和/或在轻链可变结构域中的来自“高变环”的那些残基(例如,残基26-32(li)、50-52(l2)和91-96(l3)以及在重链可变结构域中的残基26-32(hl)、53-55(142)和96-101(h3);chothia和lesk,“canonical structures for the hypervariable regions of immunoglobulins,”j.mol.biol.196:901-17(1987),其特此通过引用整体并入)。fr残基是除本文定义的高变区残基之外的那些可变结构域残基。“单链fv”或“scfv”抗体片段可以包括抗体的vh和vl结构域,其中这些结构域存在于单个多肽链中。通常,所述fv多肽进一步包含在vh和vl结构域之间的多肽连接基,其使所述scfv能够形成抗原结合所需的结构。关于scfv的综述,参见pluckthun in the pharmacologyof monoclonal antibodies,第113卷,rosenburg和moore编,springerverlag(new york 1994)第269-315页,其特此通过引用整体并入)。术语“双体”包括具有两个抗原结合位点的小抗体片段,所述片段包含在同一多肽链中连接至可变轻结构域(vl)的可变重结构域(vh)。通过使用因为太短而不允许在相同链上的两个结构域之间配对的连接基,迫使所述结构域与另一条链的互补结构域配对并建立两个抗原结合位点。双体更完整地描述在,例如,behringwerke ag的ep 0 404 097;enzon,inc.的wo 93/11161;和hollinger等人,“‘diabodies’:small bivalent and bispecificantibody fragments,”proc.natl.acad.sci.usa 90:6444-6448(1993),它们都特此通过引用整体并入。完全人抗体是有用的并且可以使用转基因小鼠生成,所述转基因小鼠不能表达内源性免疫球蛋白重链和轻链基因,但是其可以表达人重链和轻链基因。所述转基因小鼠以正常方式用选定的抗原(例如,本公开内容的多肽的全部或部分)免疫。使用常规杂交瘤技术,可以获得针对抗原的单克隆抗体。转基因小鼠所携带的人免疫球蛋白转基因在b细胞分化过程中重新排列,并随后发生类别转换和体细胞突变。因此,使用这样的技术,可能产生治疗上有用的igg、iga、igm和ige抗体。关于用于生产人抗体的这种技术的概述,参见lonberg和huszar,“human antibodies from transgenic mice,”int.rev.immunol.13:65-93(1995),其特此通过引用整体并入。关于用于生产人抗体和人单克隆抗体的该技术以及用于生产这样的抗体的方案的详细讨论,参见,例如,lonberg等人的美国专利号5,625,126;lonberg等人的美国专利号5,633,425;lonberg等人的美国专利号5,569,825;lonberg等人的美国专利号5,661,016;lonberg等人的美国专利号5,545,806,它们都特此通过引用整体并入。使用被称作“引导选择”的技术,可以产生识别选定表位的完全人抗体。在该方案中,使用选定的非人单克隆抗体(例如,小鼠抗体)引导识别相同表位的完全人抗体的选择。参见,例如,jespers等人,“guiding the selection of human antibodies from phagedisplay repertoires to a single epitope of an antigen,”biotechnology 12:899-903(1994),其特此通过引用整体并入。使用本领域已知的各种技术(包括噬菌体展示文库),也可以产生人抗体(参见,例如,hoogenboom和winter,“by-passingimmunisation.human antibodies from synthetic repertoires of germline vh genesegments rearranged in vitro,”j.mol.biol.227:381(1991);marks等人,“by-passingimmunization.human antibodies from v-gene libraries displayed on phage,”j.mol.biol.222:581(1991);quan和carter,“the rise of monoclonal antibodies astherapeutics,”见anti-ige and allergic disease,jardieu和fick,编,marcel dekker(newyork,n.y.,2002)第20章,第427-469页),它们都特此通过引用整体并入。非人(例如,啮齿动物)抗体的“人源化”形式是含有源自非人免疫球蛋白的最少序列的嵌合抗体。通常,人源化抗体是人免疫球蛋白(受体抗体),其中来自受体的高变区的残基被来自非人物种(供体抗体)(诸如具有期望的特异性、亲和力和能力的小鼠、大鼠、兔或非人灵长类动物)的高变区的残基替代。在某些情况下,人免疫球蛋白的框架区(fr)残基被对应的非人残基替代。此外,人源化抗体可以包含在受体抗体或供体抗体中未发现的残基。做出这些修饰以进一步改进抗体性能。一般而言,人源化抗体将包含基本上所有的至少一个,并且通常两个可变结构域,其中所有的或基本上所有的高变环对应于非人免疫球蛋白的高变环,且所有的或基本上所有的fr是人免疫球蛋白序列的fr。人源化抗体还任选地包含免疫球蛋白恒定区(fc)(通常为人免疫球蛋白的恒定区)的至少一部分。关于其它细节,参见jones等人,“replacing the complementarity-determining regions in a humanantibody with those from a mouse,”nature 321:522-25(1986);riechmann等人,“reshaping human antibodies for therapy,”nature 332:323-329(1988);和presta,l.“antibody engineering,”curr.op.struct.biol.2:593-596(1992),它们都特此通过引用整体并入。使用标准的重组dna技术可以制备的包含人和非人部分的重组抗体(诸如嵌合的和人源化的单克隆抗体)是有用的抗体。嵌合抗体是其中不同部分源自不同动物物种的分子,例如,具有源自鼠单克隆的可变区和人免疫球蛋白恒定区的分子(参见,例如,cabilly等人的美国专利号4,816,567;和boss等人的美国专利号4,816,397,它们通过引用整体并入本文)。人源化抗体是来自非人物种的抗体分子,其具有来自非人物种的一个或多个互补性决定区(cdr)以及来自人免疫球蛋白分子的框架区(参见,例如,queen等人的美国专利号5,585,089,其通过引用整体并入本文)。这样的嵌合的和人源化的单克隆抗体可以通过本领域已知的重组dna技术生产,例如使用在以下文献中描述的方法:int genetic eng的国际公开号wo 87/02671;teijin ltd的欧洲专利公开号0 184 187;japan res dev corp的欧洲专利公开号0 171 496;univ leland stanford junior的欧洲专利公开号0 173 494;celltech ltd的国际公开号wo 86/01533;cabilly等人的美国专利号4,816,567;berter等人,“escherichia coli secretion of an active chimeric antibody fragment,”science240:1041-1043(1988);liu等人,“chimeric mouse-human igg1antibody thatcan mediate lysis of cancer cells,”proc.natl.acad.sci.usa 84:3439-3443(1987);liu等人,“production of a mouse-human chimeric monoclonal antibody to cd20with potent fc-dependent biologic activity,”j.immunol.139:3521-3526(1987);sun等人,“chimeric antibody with human constant regions and mouse variableregions directed against carcinoma-associated antigen 17-1a,”proc.natl.acad.sci.usa 84:214-218(1987);nishimura等人,“recombinant human-mouse chimeric monoclonal antibody specific for common acute lymphocyticleukemia antigen,”cancer.res.47:999-1005(1987);wood等人,“the synthesis and invivo assembly of functional antibodies in yeast,”nature 314:446-449(1985);和shaw等人,“mouse/human chimeric antibodies to a tumor-associated antigen:biologic activity of the four human igg subclasses,”j.natl.cancer inst.80:1553-1559(1988);morrison,s.l.,“transfectomas provide novel chimericantibodies,”science 229:1202-1207(1985);winter的美国专利号5,225,539;jones等人,“replacing the complementarity-determining regions in a human antibodywith those from a mouse,”nature321:552-525(1986);verhoeyan等人,“reshapinghuman antibodies:grafting an antilysozyme activity,”science 239:1534(1988);和beidler等人,“cloning and high level expression of a chimeric antibody withspecificity for human carcinoembryonic antigen,”j.immunol.141:4053-4060(1988),它们都特此通过引用整体并入。如本文描述的,“分离的”包括与以下物质的其它组分分离:(a)天然来源,诸如植物或动物细胞或细胞培养物,或(b)合成的有机化学反应混合物。本文中使用的“纯化的”是指,当分离时,该分离物含有按该分离物的重量计至少95%、并在另一个方面至少98%的化合物(例如,缀合物)。“分离的”抗体是已经从其天然环境的组分中鉴定和分离和/或回收的抗体。其天然环境的污染组分是会干扰所述抗体的诊断或治疗用途的物质,并且可以包括酶、激素和其它蛋白性的或非蛋白性的溶质。在某些实施方案中,可以将所述抗体纯化(1)至大于95重量%的抗体(按lowry方法测定),并且在某些实施方案中超过99重量%,(2)至足以获得n-端或内部氨基酸序列的至少15个残基的程度(通过使用旋转杯序列分析仪)或(3)至同质性(使用考马斯蓝或优选银染色在还原或非还原条件下通过sds-page测定)。分离的抗体可以包括在重组细胞内的原位抗体,因为抗体的天然环境的至少一种组分将不存在。通常,分离的抗体可以通过至少一个纯化步骤来制备。“诱导细胞凋亡”的抗体是诱导程序性细胞死亡的抗体,这通过膜联蛋白v的结合、dna的片段化、细胞收缩、内质网的扩张、细胞片段化和/或膜囊泡(被称为细胞凋亡小体)的形成来确定。所述细胞可以是肿瘤细胞,例如,乳房、卵巢、胃、子宫内膜、唾液腺、肺、肾、结肠、甲状腺、胰腺或膀胱细胞。各种方法可用于评价与细胞凋亡相关的细胞事件。例如,可以通过膜联蛋白结合来测量磷脂酰丝氨酸(ps)易位;可以通过dna梯带法来评价dna片段化;并且可以通过亚二倍体细胞的任何增加来评价核/染色质凝聚以及dna片段化。在其它实施方案中,所述抗体是抗体或其功能活性片段的融合蛋白,例如其中所述抗体通过共价键(例如,肽键)在n-端或c-端融合至并非来自抗体的另一种蛋白(或其部分,优选所述蛋白的至少10、20或50个氨基酸部分)的氨基酸序列。在一个实施方案中,所述抗体或其片段在恒定结构域的n-端处共价连接至另一种蛋白。抗体包括类似物和衍生物,其经过修饰,即,通过共价连接任何类型的分子,只要这样的共价连接允许所述抗体保留其抗原结合免疫特异性。例如,抗体的衍生物和类似物包括已经进一步修饰的那些,例如,通过糖基化、乙酰化、聚乙二醇化、磷酸化、酰胺化、通过已知的保护/封闭基团的衍生化、蛋白水解性裂解、与细胞抗体单元或其它蛋白的连接。多种化学修饰中的任一种可以通过已知技术进行,包括、但不限于特异性化学裂解、乙酰化、甲酰化和在有衣霉素存在下的代谢合成。另外,所述类似物或衍生物可以含有一种或多种非天然氨基酸。抗体可以具有在与fc受体相互作用的氨基酸残基中的修饰(例如,置换、缺失或添加)。具体地,抗体可以具有在被鉴定为参与抗-fc结构域和fcrn受体之间的相互作用的氨基酸残基中的修饰(参见,例如,国际公开号wo 97/34631,其通过引用整体并入本文)。在一个实施方案中,ab(即,抗体)是肿瘤靶向抗体、抗体片段、双特异性抗体或抗体片段、单克隆抗体、嵌合抗体或人源化抗体。对癌细胞抗原免疫特异性的抗体可以商购得到,或通过本领域技术人员已知的任何方法(例如,化学合成或重组表达技术)生产。编码对癌细胞抗原免疫特异性的抗体的核苷酸序列可以得自例如genbank数据库或类似于它的数据库、文献出版物,或通过常规克隆和测序得到。在一个实施方案中,ab(即,抗体)选自由以下成员组成的集合:抗-her2抗体、抗-cd20抗体、抗-cd38抗体、抗-il-6受体抗体、抗-vegrf2抗体、抗-her-2抗体、抗-dll3抗体、抗-连接素4抗体、抗-cd33抗体、抗-cd79b抗体、抗-cd11a抗体、抗-bcma抗体、抗-cd22抗体、抗-trop2抗体、抗-frα抗体、抗-epcam抗体、抗-间皮素抗体、抗-liv1抗体、奥戈伏单抗、依决洛单抗、西妥昔单抗、针对玻璃体结合蛋白受体(αvβ3)的人源化单克隆抗体、阿仑珠单抗、用于治疗非霍奇金淋巴瘤的人源化抗-hla-dr抗体、131l lym-1、用于治疗非霍奇金淋巴瘤的鼠抗-hla-drl0抗体、用于治疗霍奇金病或非霍奇金淋巴瘤的人源化抗-cd2mab、拉贝珠单抗、贝伐珠单抗、替伊莫单抗、奥法木单抗、帕尼单抗、利妥昔单抗、托西莫单抗、伊匹木单抗、吉妥珠单抗、针对癌胚蛋白受体5t4的的人源化单克隆抗体、m1/70(针对cd11b受体的抗体)、抗-mrc1、抗gcc、抗cd32和其它抗体。在一个实施方案中,可以使用用于治疗癌症的已知抗体。对癌细胞抗原免疫特异性的抗体可以商购得到,或通过本领域技术人员已知的任何方法(例如,重组表达技术)生产。编码对癌细胞抗原免疫特异性的抗体的核苷酸序列可以得自例如genbank数据库或类似于它的数据库、文献出版物,或通过常规克隆和测序得到。可用于治疗癌症的抗体的例子包括、但不限于:奥戈伏单抗或其为用于治疗卵巢癌的鼠抗体;依决洛单抗或panorex,其为用于治疗结肠直肠癌的鼠igg2a抗体;西妥昔单抗(例如,),其为用于治疗表皮生长因子阳性的癌症(诸如头颈癌)的抗-egfr igg嵌合抗体;vitaxin,其为用于治疗肉瘤的人源化抗体;阿仑珠单抗或campath-1h,其为用于治疗慢性淋巴细胞性白血病(cll)的人源化igg1抗体;oncolym,其为用于治疗非霍奇金淋巴瘤的放射性标记的鼠抗-hla-dr10抗体;allomune(bio transplant,ca),其为用于治疗霍奇金病或非霍奇金淋巴瘤的人源化抗-cd2 mab;和cea-cide(immunomedics,nj),其为用于治疗结肠直肠癌的人源化抗-cea抗体。术语“蛋白”、“多肽”和“肽”在本文中可以互换使用。这些术语可以如下区分。蛋白通常表示细胞中转录、翻译和翻译后修饰的最终产物。多肽可以包括蛋白或肽。与蛋白相反,肽通常是氨基酸的短聚合物,其长度通常为100个或更少的氨基酸。本文中使用的术语“肽”或“多肽”表示蛋白及其片段。肽可以包括氨基酸序列。可以以从氨基端到羧基端的方向从左到右书写那些序列。根据标准命名法,氨基酸残基序列由三字母或单字母代码命名,如下面所示:丙氨酸(ala,a),精氨酸(arg,r),天冬酰胺(asn,n),天冬氨酸(asp,d),瓜氨酸(cit),半胱氨酸(cys,c),谷氨酰胺(gln,q),谷氨酸(glu,e),甘氨酸(gly,g),组氨酸(his,h),异亮氨酸(ile,i),亮氨酸(leu,l),赖氨酸(lys,k),甲硫氨酸(met,m),苯丙氨酸(phe,f),脯氨酸(pro,p),丝氨酸(ser,s),苏氨酸(thr,t),色氨酸(trp,w),酪氨酸(tyr,y),和缬氨酸(val,v)。所述免疫治疗化合物的肽可以源自天然的,或者可以可替换地重新设计。如果肽可以通过片段化天然存在的序列来获得,或者如果肽可以基于天然存在的氨基酸序列或编码该序列的遗传物质(dna或rna)的序列的知识来合成,就说所述肽“可衍生自天然存在的氨基酸序列”。所述免疫治疗化合物的肽可以与或可以不与天然存在的蛋白或其部分(例如,肽)具有实质同源性或同一性。所述免疫治疗化合物可以包括或不包括与天然存在的蛋白或其部分(例如,肽)具有“实质相似性”的肽。具有实质相似性的肽包括与特定肽具有至少70%或更大序列同源性或同一性的肽,所述特定肽具有与参考肽相同数目的氨基酸残基。术语载荷或“药物载荷”或“有效负载载荷”表示在adc分子中每个抗体的有效负载(“有效负载”在本文中与“药物”可互换地使用)的平均数目。药物载荷可以在每个抗体1至50个药物的范围内。这有时被称作dar,或药物与抗体的比率。本文描述的adc的组合物通常具有1-25的dar,并且在某些实施方案中,具有1-8、2-8、2-6、2-5和2-4的dar。典型的dar值包括2、4、6、8和10。通过常规方式诸如紫外/可见光光谱法、质谱法、elisa测定和hplc,可以表征每个抗体的平均药物数目或dar值。还可以确定定量dar值。在某些情况下,具有特定dar值的同质adc的分离、纯化和表征可以通过诸如反相hplc或电泳等方式来实现。dar可能受限于抗体上连接位点的数目。例如,在所述连接是半胱氨酸硫醇的情况下,抗体可以仅具有一个或几个半胱氨酸硫醇基团,或者可以仅具有一个或几个可连接连接基单元的足够反应性的硫醇基团。在某些实施方案中,所述半胱氨酸硫醇是形成链间二硫键的半胱氨酸残基的硫醇基团。在某些实施方案中,所述半胱氨酸硫醇是不形成链间二硫键的半胱氨酸残基的硫醇基团。通常,在缀合反应期间与抗体缀合的药物部分少于理论最大值。抗体可以含有例如许多不与连接基或连接基中间体反应的赖氨酸残基。只有最有反应性的赖氨酸基团可以与反应性连接基试剂反应。通常,抗体不含有许多(如果存在的话)游离的且反应性的半胱氨酸硫醇基团,其可以通过连接基与药物连接。在抗体中的大多数半胱氨酸硫醇残基作为二硫键存在,并且必须用还原剂诸如二硫苏糖醇(dtt)还原。所述抗体可以经历变性条件以揭示反应性亲核基团诸如赖氨酸或半胱氨酸。adc的载荷(药物/抗体比率)可以通过几种不同的方式控制,包括:(i)限制药物-连接基相对于抗体的摩尔过量,(ii)限制缀合反应时间或温度,和(iii)半胱氨酸硫醇修饰的部分或限制性还原条件。在超过一个亲核基团与药物-连接基反应的情况下,则所得产物是每个抗体分布有一个或多个药物部分的adc的混合物。每个抗体的平均药物数目可以从混合物计算,例如通过对抗体特异性的和对药物特异性的双elisa抗体测定。在混合物中的各种adc可以通过质谱法进行鉴定,并通过hplc(例如,疏水相互作用色谱法)进行分离。在一个实施方案中,所述抗体可以选自曲妥珠单抗和曲妥珠单抗突变体。在某些实施方案中,所述抗体通过以下物质而结合:用酰基供体含谷氨酰胺标签(例如,含gln的肽标签或q-标签)工程改造的含fc的或含fab的多肽,或通过多肽工程(例如,通过在多肽上的氨基酸缺失、插入、置换、突变或它们的任何组合)变得在有转谷氨酰胺酶存在下具有反应性(即,在有胺和转谷氨酰胺酶存在下形成共价键作为酰基供体的能力)的内源性谷氨酰胺。在某些实施方案中,本公开内容涉及前述抗体药物缀合物和附带定义中的任一种,其中所述抗体药物缀合物包含2、3、4、5、6、7、8、9、10、15、20或25种本公开内容的化合物或其中任意数目的化合物。在某些实施方案中,本公开内容涉及前述抗体药物缀合物和附带定义中的任一种,其中所述抗体药物缀合物包含3或4种本公开内容的化合物。氨基酸“衍生物”包括通过母体氨基酸的共价连接(诸如,例如,通过烷基化、糖基化、乙酰化、磷酸化等)而具有取代或修饰的氨基酸。在“衍生物”的预期含义内进一步包括例如具有取代的连接的氨基酸的一种或多种类似物,以及本领域已知的其它修饰。“天然的氨基酸”表示精氨酸、谷氨酰胺、苯丙氨酸、酪氨酸、色氨酸、赖氨酸、甘氨酸、丙氨酸、组氨酸、丝氨酸、脯氨酸、谷氨酸、天冬氨酸、苏氨酸、半胱氨酸、甲硫氨酸、亮氨酸、天冬酰胺、异亮氨酸和缬氨酸,除非上下文另外指出。连接基(在本文中有时被称作“[连接基]”)是可以用于连接药物和抗体以形成抗体药物缀合物(adc)的双功能化合物。这样的缀合物可用于例如形成针对肿瘤相关抗原的免疫缀合物。在某些实施方案中,这样的缀合物可以允许将细胞毒性药物选择性递送至肿瘤细胞。本文描述的自降解的间隔基包括共价组装体,其被定制为与在前体中的保护性部分被活化以后两个化学键的切割相关联:在受到刺激后,保护性部分被除去,这会产生一系列分解反应,从而导致较小分子在时间上按顺序释放。参见alouane等人,“self-immolative spacers:kinetic aspects,structure-property relationships,andapplications,”angewandte chemie 54(26):7492-7509(2015),其特此通过引用整体并入。自降解的间隔基的创造是为了解决药物递送的限制,并已经在药物化学、分析化学和材料科学中引起了广泛兴趣。参见alouane等人,“self-immolative spacers:kineticaspects,structure-property relationships,and applications,”angewandte chemie54(26:7492-7509(2015),其特此通过引用整体并入。短语“实质量”包括混合物或样品的大多数,即群体中的大于50%。术语“细胞内代谢物”表示在细胞内对抗体-药物缀合物(adc)的代谢过程或反应所产生的化合物。所述代谢过程或反应可以是酶促过程,诸如adc的肽连接基的蛋白水解性裂解。细胞内代谢物包括、但不限于在进入、扩散、摄取或运输到细胞中以后已经经历细胞内切割的抗体和游离药物。术语“细胞内地切割”和“细胞内切割”表示在细胞内对adc等的代谢过程或反应,由此在药物部分和所述抗体之间的共价连接(例如连接基)被破坏,从而产生游离的药物或在细胞内与抗体分离的缀合物的其它代谢物。因此,adc的被切割部分是细胞内代谢物。术语“生物利用度”表示施用给患者的给定量的药物的全身利用度(即,血液/血浆水平)。生物利用度指示药物从施用的剂型到达全身循环的时间(速率)和总量(程度)的测量。术语“细胞毒性活性”表示adc或所述adc的细胞内代谢物的细胞杀死作用、细胞生长抑制作用或抗增殖作用。细胞毒性活性可以表示为ic50值,其为一半细胞存活时每单位体积的浓度(摩尔或质量)。“障碍”是将受益于药物或抗体-药物缀合物治疗的任何病症。这包括慢性和急性障碍或疾病,包括使哺乳动物易患所讨论的障碍的那些病理学病症。在本文中要治疗的障碍的非限制性例子包括良性和恶性癌症;白血病和淋巴样恶性肿瘤、神经元障碍、神经胶质障碍、星形细胞障碍、下丘脑和其它腺体障碍、巨噬细胞障碍、上皮障碍、间质障碍和囊胚腔障碍;以及炎症性障碍、血管生成性障碍和免疫学障碍。术语“癌症”和“癌性的”表示或描述哺乳动物中的生理病症或障碍,其通常特征在于失调的细胞生长。“肿瘤”包含一种或多种癌细胞。本文描述的感染性疾病包括任何病毒感染、细菌感染、真菌感染或它们的任何组合。根据本文描述的方法可以治疗的示例性病毒感染包括、但不限于冠状病毒(例如,严重急性呼吸综合征(sars)、sars-cov-2(covid-19)、中东呼吸综合征(mers)和普通感冒)、埃博拉、流感、肝炎、hib疾病、人免疫缺陷病毒(hiv)、人乳头瘤病毒(hpv)、脑膜炎球菌疾病、肺炎球菌疾病、麻疹、腮腺炎、诺如病毒、脊髓灰质炎、呼吸道合胞病毒(rsv)、轮状病毒、风疹病毒、带状疱疹、西尼罗河病毒、狂犬病病毒、肠道病毒、巨细胞病毒、疱疹病毒、水痘、黄热病、寨卡病毒或它们的任何组合。根据本文描述的方法可以治疗的示例性细菌感染包括、但不限于链球菌疾病、葡萄球菌疾病、白喉、脑膜炎球菌疾病、破伤风、百日咳、肺炎球菌疾病、细菌食品中毒、性传播的感染、结核病、莱姆病、肉毒中毒或它们的任何组合。根据本文描述的方法可以治疗的示例性真菌感染包括、但不限于念珠菌病、组织胞浆菌病、皮肤癣菌病、足癣、曲霉菌病、隐球菌性脑膜炎、球孢子菌病或它们的任何组合。本文中使用的术语“细胞”、“细胞系”和“细胞培养”可互换使用并且所有这样的命名包括后代。词语“转化体”和“转化的细胞”包括原代受试者细胞和由其衍生出的培养物或后代,而不考虑转移的次数。还理解,由于故意的或非故意的突变,所有后代可以在dna含量上不是精确地相同的。包括具有相同功能或生物活性的突变体后代,如在最初转化的细胞中所筛选的。在意指不同名称时,它将从上下文中显而易见。本文中使用的“患者”包括人类和其它动物,特别是哺乳动物。因此,所述方法适用于人治疗和兽医应用。“患者”的例子包括、但不限于人、大鼠、小鼠、豚鼠、猴、猪、山羊、牛、马、狗、猫、鸟和家禽。在某些实施方案中,所述患者是哺乳动物,例如,灵长类动物。在某些实施方案中,所述患者是人。在一个实施方案中,所述患者是婴儿、少年或成人。除非上下文另外指出,否则术语“治疗”表示治疗性治疗和预防复发的预防性措施,其中目的是抑制或减慢(减轻)不希望的生理学变化或障碍,诸如癌症或病毒感染的发展或扩散。为了本公开内容的目的,有益的或期望的临床结果包括、但不限于:症状的减轻、疾病程度的减小、疾病的稳定化(即不恶化)状态、疾病进展的推迟或减慢、疾病状态的改善或缓和、以及缓解(无论是部分的还是完全的),无论是可检测的还是不可检测的。“治疗”也可以意指与如果不接受治疗所预期的存活期相比延长的存活。需要治疗的受试者包括已经具有病症或障碍的那些以及易于具有病症或障碍的那些。在癌症的上下文中,术语“治疗”包括以下中的任一种或全部:抑制肿瘤细胞、癌细胞或肿瘤的生长;抑制肿瘤细胞或癌细胞的复制;减轻总体肿瘤负荷或减少癌细胞的数目;以及改善与该疾病相关的一种或多种症状。在自身免疫性疾病的上下文中,术语“治疗”包括以下中的任一种或全部:抑制与自身免疫性疾病状态相关的细胞(包括、但不限于产生自身免疫性抗体的细胞)的复制,减轻自身免疫性抗体负担,以及改善自身免疫性疾病的一种或多种症状。在感染性疾病的上下文中,术语“治疗”包括以下中的任一种或全部:抑制造成感染性疾病的病原体的生长、繁殖或复制,以及改善感染性疾病的一种或多种症状。治疗可以涉及向诊断出患有疾病的患者施用本文描述的化合物,并且可以涉及向不具有活动性症状的患者施用所述化合物。相反,治疗可以涉及向处于发生特定疾病的风险中的患者或向报告疾病的一种或多种生理学症状的患者施用所述组合物,即使可能尚未做出该疾病的诊断。关于本发明剂型的术语“施用”表示将所述剂型引入需要治疗的受试者的系统中的行为。当本发明的剂型与一种或多种其它活性剂(在其各自的剂型中)组合施用时,“施用”及其变体各自被理解为包括同时和/或相继引入所述剂型和其它活性剂。所描述的任何剂型的施用包括平行施用、共同施用或依次施用。在某些情况下,大约同时施用所述疗法,例如彼此相隔约几秒至几小时。本文描述的化合物的“治疗上有效的”量通常是足以实现所需效果的量,并且可以根据疾病状况的性质和严重程度以及所述化合物的效能而变化。应当理解,可以采用与活动性疾病的治疗不同的浓度来预防。治疗益处如下实现:改善与根本障碍相关的一种或多种生理学症状,从而在患者中观察到改善,尽管患者可能仍受潜在障碍折磨。就癌症而言,治疗有效量的药物可以减少癌细胞的数目;减小肿瘤大小;抑制(即,在某种程度上减慢和优选地停止)癌细胞向周围器官中的浸润;抑制(即,在某种程度上减慢和优选地停止)肿瘤转移;在某种程度上抑制肿瘤生长;和/或在某种程度上缓解与癌症相关的一种或多种症状。在所述药物可以抑制现有癌细胞的生长和/或杀死现有癌细胞的范围内,它可能是细胞生长抑制性的和/或细胞毒性的。对于癌症治疗而言,例如通过评估达到疾病进展的时间(ttp)和/或确定应答率(rr),可以测量效力。因此,治疗效果可以是与障碍相关的症状的严重程度的降低和/或障碍进展的抑制(部分的或完全的),或障碍或副作用的改善的治疗、治愈、预防或消除。基于受试者的年龄、健康、大小和性别,可以确定引发治疗应答所需的量。也可以基于受试者对治疗的应答的监测来确定最佳量。术语“治疗”可以包括有效抑制、遏制或停止症状,从而预防或延迟病症的发作、延缓病症的进展或改善病症的症状。贯穿本说明书,术语和取代基保留其定义。取代基(例如,rn)通常在引入时被定义,并贯穿说明书和在所有独立权利要求中保留该定义。c1至c20烃包括含有1至20个碳原子(包括端值)的烷基、环烷基、多环烷基、烯基、炔基、芳基、及其组合。非限制性例子包括乙基、苄基、苯乙基、环己基甲基、樟脑基和萘基乙基。烃表示由氢和碳作为仅有的元素组分组成的任何取代基。烷基是烃的一个子集。除非另外指出,否则烷基(或亚烷基)意图包括直链或支链饱和烃结构及其组合。在某些实施方案中,烷基表示1至20个碳原子、或1至10个碳原子、或1至8个碳原子、或1至6个碳原子、或1至5个碳原子、或1至4个碳原子的烷基基团。烷基基团的例子包括甲基、乙基、丙基、异丙基、正丁基、仲丁基、叔丁基等。环烷基是烃的一个子集,并且包括3至8个碳原子的环状烃基。环烷基基团的例子包括环丙基、环丁基、环戊基、降冰片基等。氧杂烷基表示其中一个或多个碳(及其相关的氢)已经被氧替换的烷基残基。例子包括甲氧基丙氧基、羟甲基、羟乙基、3,6,9-三氧杂癸基等。术语氧杂烷基如本领域所理解的那样意指[参见naming and indexing of chemical substances for chemical abstracts,american chemical society出版,196,但不受127(a)的限制],即它表示其中氧通过单键与其相邻原子键合(形成醚键)的化合物;它不表示双键氧,如在羰基基团中所见的。类似地,氮杂烷基表示其中一个或多个碳已经被氮替换的烷基残基。例子包括乙基氨基乙基和甲基氨基丙基等。烷氧基(alkoxy或alkoxyl)是氧杂烷基的一个子集,并且表示通过氧连接至母体结构的1至20个碳原子的基团。在某些实施方案中,烷基表示1至20个碳原子、或1至10个碳原子、或1至6个碳原子、或1至5个碳原子、或1至4个碳原子的具有直链或支链构型的烷基基团。例子包括甲氧基、乙氧基、丙氧基、异丙氧基等。低级烷氧基表示含有一至四个碳的基团。为了本技术的目的,烷氧基和低级烷氧基包括亚甲基二氧基和亚乙基二氧基。芳基和杂芳基是指(i)苯基基团(或苯)或含有1-4个选自o、n或s的杂原子的单环5或6元杂芳族环;(ii)含有0-4个选自o、n或s的杂原子的二环9或10元芳族或杂芳族环系统;或(iii)含有0-5个选自o、n或s的杂原子的三环13或14元芳族或杂芳族环系统。芳族6-14元碳环包括例如苯、萘、茚满、四氢萘和芴,且5-10元芳族杂环包括例如咪唑、吡啶、吲哚、噻吩、苯并吡喃酮、噻唑、呋喃、苯并咪唑、喹啉、异喹啉、喹喔啉、嘧啶、吡嗪、四唑和吡唑。本文中使用的芳基和杂芳基表示其中一个或多个环是芳族、但不需要都是芳族的残基。在某些实施方案中,芳基表示苯基基团。在某些实施方案中,杂芳基表示吡啶、咪唑、嘧啶、吲哚、噻吩、苯并吡喃酮、噻唑、呋喃、苯并咪唑、喹啉、异喹啉、喹喔啉、嘧啶、吡嗪、四唑和吡唑。在其它实施方案中,杂芳基表示吡啶、哒嗪、吡嗪或嘧啶。在再其它的实施方案中,杂芳基表示吡啶。杂环是指其中1至4个碳被选自n、o和s的杂原子替换的环烷基或芳基碳环残基。氮和硫杂原子可以任选地被氧化,并且氮杂原子可以任选地被季铵化。除非另外指出,否则杂环可以是非芳族的或芳族的。落入本发明范围内的杂环的非限制性例子包括吡咯烷、吡唑、吡咯、吲哚、喹啉、异喹啉、四氢异喹啉、苯并呋喃、苯并二氧杂环己烷、苯并二氧杂环戊烯(当作为取代基出现时,通常被称作亚甲基二氧基苯基)、四唑、吗啉、噻唑、吡啶、哒嗪、嘧啶、噻吩、呋喃、噁唑、噁唑啉、异噁唑、二氧杂环己烷、四氢呋喃等。应当指出,杂芳基是杂环的一个子集,其中所述杂环是芳族的。杂环基残基的例子另外包括哌嗪基、2-氧代哌嗪基、2-氧代哌啶基、2-氧代-吡咯烷基、2-氧代氮杂环庚三烯基、氮杂环庚三烯基、4-哌啶基、吡唑烷基、咪唑基、咪唑啉基、咪唑烷基、吡嗪基、噁唑烷基、异噁唑烷基、噻唑烷基、异噻唑基、奎宁环基、异噻唑烷基、苯并咪唑基、噻二唑基、苯并吡喃基、苯并噻唑基、四氢呋喃基、四氢吡喃基、噻吩基、苯并噻吩基、硫吗啉基、硫吗啉基亚砜、硫吗啉基砜、噁二唑基、三唑基和四氢喹啉基。氮杂环是在环中含有至少一个氮的杂环;它可能含有另外的氮以及其它杂原子。非限制性例子包括哌啶、哌嗪、吗啉、吡咯烷和硫代吗啉。氮杂芳基是氮杂环一个子集;例子包括吡啶、吡咯和噻唑。术语“卤素”是指氟、氯、溴或碘原子。在一个实施方案中,卤素可以是氟或氯原子。除非另外指出,否则酰基表示甲酰基以及通过羰基官能团连接至母体结构的具有1、2、3、4、5、6、7和8个碳原子的直链、支链、环状构型、饱和的、不饱和的和芳族的及其组合的基团。例子包括乙酰基、苯甲酰基、丙酰基、异丁酰基等。低级酰基表示含有一至四个碳的基团。双键氧当被称作取代基本身时被称为“氧代”。本文中使用的术语“任选地被取代的”可以与“未被取代的或被取代的”互换使用。术语“取代”表示用指定残基替换指定基团中的一个或多个氢原子。例如,“被取代的芳基”或“被取代的杂芳基”表示这样的芳基或杂芳基,其中在每个残基中的一个或多个h原子被卤素、卤代烷基、烷基、烷氧基或卤代烷氧基替换。本文描述的化合物可以含有不对称的中心且因此可能产生对映异构体、非对映异构体和其它立体异构形式,它们可以以绝对立体化学的方式定义为(r)-或(s)-。本发明意在包括所有这样的可能的非对映异构体以及它们的外消旋的和光学纯的形式。光学活性的(r)-和(s)-异构体可以使用同手性合成子或同手性试剂来制备,或者使用常规技术进行光学拆分。当本文所述的化合物含有烯族双键或其它几何不对称中心时,并且除非另有说明,否则它意图包括(e)-和(z)-几何异构体。同样地,意图包括所有互变异构形式。本文所用的外消旋的、ambiscalemic和scalemic或对映异构纯的化合物的图形表示是取自maehr j.chem.ed.62,114-120(1985)的表示的修改版本:简单线不提供关于立体化学的信息,并且仅传达连接性;实心楔和虚形楔用于表示手性元素的绝对构型;实和虚粗线是几何描述信息,指示所示的相对构型,但不一定表示外消旋特征;且楔形轮廓和点状或虚线表示不确定绝对构型的对映异构纯的化合物。例如,图形表示指示两种反式:反式对映异构体中的任一种或两种:其以任何比例,从纯的对映异构体到外消旋体。图形表示:指示未知绝对立体化学的单一对映异构体,即,它可以是前述两种结构中的任一种,作为基本上纯的单一对映异构体。并且最后,图形表示:指示纯的(r,r,s)绝对构型。为了本公开内容的目的,“纯的”或“基本上纯的”对映异构体意图指,所述对映异构体是至少95%的所示构型以及5%或更少的其它对映异构体。类似地,“纯的”或“基本上纯的”非对映异构体意图指,所述非对映异构体是至少95%的所示相对构型以及5%或更少的其它非对映异构体。在某些实施方案中,所述化合物的纯度是至少99%。在这些可能性中的任一种中,化合物可以是单一立体异构体或混合物。如果是混合物,则该混合物将最通常是外消旋的,但它不一定是。生物活性化合物(诸如本文描述的那些)的基本上纯的单一立体异构体经常表现出优于它们的外消旋混合物的优点。对映异构纯的是指大于80e.e.,且优选地大于90e.e.。为了本公开内容的目的,“纯的”或“基本上纯的”立体异构体意图指,所述立体异构体是至少95%的所示构型和5%或更少的其它立体异构体,或至少97%的所示构型和3%或更少的其它立体异构体,或至少99%的所示构型和1%或更少的其它立体异构体。在审查后可能发现,本技术的发明人不能获得某些物种和属的专利权。在这种情况下,在申请人的权利要求中对物种和属的排除应被视为专利审查的人为因素,而不反映发明人对其发明的概念或描述,所述发明涵盖不属于公众财产的i类中的所有成员。如本文中使用的和如本领域技术人员将理解的,除非明确地进一步限制,否则“化合物”的叙述意图包括该化合物的盐。在一个特定实施方案中,术语“式……的化合物”表示所述化合物或其药学上可接受的盐。术语“药学上可接受的盐”表示从药学上可接受的无毒的酸或碱(包括无机酸和碱以及有机酸和碱)制备的盐。当本发明的化合物是碱性时,可以从药学上可接受的无毒的酸(包括无机酸和有机酸)制备盐。适用于本发明化合物的药学上可接受的酸加成盐包括乙酸、己二酸、海藻酸、抗坏血酸、天冬氨酸、苯磺酸(苯磺酸盐)、苯甲酸、硼酸、丁酸、樟脑酸、樟脑磺酸、碳酸、柠檬酸、乙烷二磺酸、乙磺酸、乙二胺四乙酸、甲酸、富马酸、葡萄庚糖酸、葡糖酸、谷氨酸、氢溴酸、盐酸、氢碘酸、羟基萘甲酸、羟乙磺酸、乳酸、乳糖酸、月桂基磺酸、马来酸、苹果酸、扁桃酸、甲磺酸、粘酸、亚萘基磺酸、硝酸、油酸、扑酸、泛酸、磷酸、新戊酸、聚半乳糖醛酸、水杨酸、硬脂酸、琥珀酸、硫酸、鞣酸、酒石酸、teoclatic、对甲苯磺酸等。当所述化合物含有酸性侧链时,本发明的化合物的合适的药学上可接受的碱加成盐包括、但不限于由铝、钙、锂、镁、钾、钠和锌制成的金属盐,或者由赖氨酸、精氨酸、n,n’-二苄基乙二胺、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、葡甲胺(n-甲基还原葡糖胺)和普鲁卡因制成的有机盐。其它药学上可接受的盐包括,在适当时,无毒的铵阳离子和与具有1-20个碳原子的烷基连接的羧酸根、磺酸根和膦酸根阴离子。本文还提供了一种药物组合物,其包含上文公开的化合物或其药学上可接受的盐形式,以及药学上可接受的载体、稀释剂或赋形剂。虽然本文公开的化合物可能作为化学原料施用,但优选将它们作为药物组合物提供。根据另一个方面,本发明提供了一种药物组合物,其包含式i的化合物或其药学上可接受的盐,以及一种或多种其药学载体和任选的一种或多种其它治疗成分。所述载体必须是“可接受的”,其含义是,与制剂的其它成分相容,且对其接受者无害。在一个实施方案中,所述药学上可接受的载体选自液体填充剂、固体填充剂、稀释剂、赋形剂、溶剂和包封材料。药学上可接受的载体(例如,添加剂诸如稀释剂、免疫刺激剂、辅助剂、抗氧化剂、防腐剂和增溶剂)对于在所采用的剂量和浓度暴露于它的细胞或受试者是无毒的。药学上可接受的载体的例子包括水,例如,用磷酸盐、柠檬酸盐和另一种有机酸缓冲的水。在本公开内容中可能有用的药学上可接受的赋形剂的代表性例子包括抗氧化剂诸如抗坏血酸;低分子量(小于约10个残基)多肽;蛋白,诸如血清白蛋白、明胶或免疫球蛋白;辅助剂(经过选择从而避免辅助剂诱导的毒性,诸如在美国专利号6,355,625(特此通过引用整体并入)中描述的3-葡聚糖,或粒细胞集落刺激因子(gcsf));亲水聚合物诸如聚乙烯吡咯烷酮;氨基酸诸如甘氨酸、谷氨酰胺、天冬酰胺、精氨酸或赖氨酸;单糖、二糖和其它碳水化合物,包括葡萄糖、甘露糖或糊精;螯合剂诸如edta;糖醇诸如甘露醇或山梨醇;形成盐的抗衡离子诸如钠;和/或非离子表面活性剂诸如聚乙二醇(peg)和在一个实施方案中,所述组合物可以进一步包含辅助剂。合适的辅助剂是本领域已知的且包括、而不限于鞭毛蛋白、弗氏完全或不完全辅助剂、氢氧化铝、溶血卵磷脂、普朗尼克多元醇、聚阴离子、肽、油乳剂、二硝基苯酚、iscomatrix和脂质体聚阳离子dna颗粒。所述制剂包括适合胃肠外(包括皮下、真皮内、肌肉内、静脉内和关节内)、直肠和局部(包括真皮、含服、舌下和眼内)施用的制剂。最合适的途径可能取决于接受者的病症和障碍。所述制剂可以方便地以单位剂型存在,并且可以通过药学领域众所周知的任意方法来制备。所有方法包括以下步骤:使本文公开的化合物或其药学上可接受的盐(“活性成分”)与构成一种或多种助剂的载体结合。一般而言,如下制备制剂:使所述活性成分与液体载体或精细粉碎的固体载体或两者均匀地且紧密地结合,然后如果必要,将产物成形为需要的制剂。用于胃肠外施用的制剂包括水性的和非水性的无菌注射溶液,其可以含有抗氧化剂、缓冲剂、抑菌剂和使制剂与预期接受者的血液等渗的溶质。用于胃肠外施用的制剂还包括水性的和非水性的无菌混悬液,其可以包括助悬剂和增稠剂。所述制剂可以存在于多剂量容器的单位剂量中,例如密封的安瓿和管形瓶,并且可以在冷冻干燥(低压冻干)条件下储存,仅需要在即将使用前添加无菌液体载体,例如盐水、磷酸盐-缓冲盐水(pbs)等。可以从前述种类的无菌粉剂、颗粒剂和片剂制备即时注射溶液和混悬液。术语“包装说明书”用于表示治疗性产品的商业包装中通常包括的指令,其含有关于使用这样的治疗产品的适应症、用法、剂量、施用、禁忌症和/或警告的信息。应当认识到,本发明的化合物可以以放射性地标记的形式存在,即,所述化合物可以含有一个或多个其原子质量或质量数不同于在自然界中通常发现的原子质量或质量数的原子。氢、碳、磷、氟和氯的放射性同位素分别包括2h、3h、13c、14c、15n、35s、18f和36cl。含有那些放射性同位素和/或其它原子的其它放射性同位素的化合物是在本发明的范围内。氚化的(即3h)和碳-14(即,14c)放射性同位素由于其易于制备和可检测性而特别优选。含有同位素11c、13n、15o和18f的化合物非常适合用于正电子发射断层摄影术。本发明的放射性地标记的式i的化合物及其前药通常可以通过本领域技术人员众所周知的方法来制备。方便地,通过用容易得到的放射性地标记的试剂替换非放射性地标记的试剂,通过执行在实施例和方案中公开的程序,可以制备这样的放射性地标记的化合物。化合物的制备可以涉及各种化学基团的保护和去保护。本领域技术人员可以容易地确定对保护和去保护的需要以及适当保护基团的选择。在化学领域的标准教科书中讨论了用于该目的的合适基团,诸如t.w.greene和p.g.m.wuts的protective groups inorganic synthesis[john wiley&sons,new york,1999],protecting group chemistry,第1版,oxford university press,2000;以及march’s advanced organic chemistry:reactions,mechanisms,and structure,第5版,wiley-interscience publication,2001。实施例实施例1:1-(4-(氨基甲基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(e66)的制备方案1.试剂:(i)net3,ch2cl2;(ii)zn,hcoonh4,meoh;(iii)c4h9cocl.net3,thf;(iv)naoh,etoh;(v)mcpba,chcl3;(vi)异氰酸苯甲酰酯,ch2cl2;naoh,meoh(vii)20%tfa,dcm,rt,2h.步骤1.(4-(((3-硝基喹啉-4-基)氨基)甲基)苄基)氨基甲酸叔丁酯:将三乙胺(72.8mg,100μl,1.5当量,719μmol)和(4-(氨基甲基)苄基)氨基甲酸叔丁酯(136mg,1.2当量,575μmol)加入4-氯-3-硝基喹啉(100mg,1当量,479μmol)在二氯甲烷(2ml)中的溶液中。将反应混合物在40℃回流45分钟。通过uplc监测反应进程。产物(在2.83min)立即开始形成。在45min内实现反应物向期望产物的完全转化。将水加入残余物中并将产物用ch2cl2(3×)萃取。将萃取物用水洗涤,经mgso4干燥,并在真空中蒸发以得到作为亮黄色固体的期望的化合物(195.8mg,约100%)。[可替换地,将在反应之后形成的黄色沉淀物过滤,用水洗涤并在真空下干燥以得到目标化合物。]步骤2.(4-(((3-氨基喹啉-4-基)氨基)甲基)苄基)氨基甲酸叔丁酯:不经进一步纯化,将来自步骤1的物质((4-(((3-硝基喹啉-4-基)氨基)甲基)苄基)氨基甲酸叔丁酯,100mg,1当量,245μmol)在meoh(2.2ml)中的悬浮液用锌粉(80.0mg,5当量,1.22mmol)和甲酸铵(77.2mg,5当量,1.22mmol)处理。将反应混合物在室温搅拌20min以产生灰色溶液。通过uplc监测反应进展。产物立即开始形成。结束后,将反应混合物穿过硅藻土过滤并将溶剂在真空中蒸发。将残余物溶解在水中,用etoac(3×20ml)萃取,用水洗涤并经mgso4干燥。在真空下除去溶剂以得到82.2mg(88.7%)的期望的化合物,为粘性的蓬松的亮黄色物质。步骤3.(4-(((3-戊烷酰氨基喹啉-4-基)氨基)甲基)苄基)氨基甲酸叔丁酯:向在无水etoac(40ml)中的步骤2的粗产物((4-(((3-氨基喹啉-4-基)氨基)甲基)苄基)氨基甲酸叔丁酯,41.1mg,1当量,109μmol)中加入三乙胺(14.3mg,19.7μl,1.3当量,141μmol)和戊酰氯(14.4mg,14.2μl,1.1当量,119μmol)。将反应混合物回流30min。通过uplc监测反应。期望产物在30分钟内从溶液中沉淀。将溶剂在减压下蒸发并将残余物溶解在etoac中,并用水洗涤。将有机级分经mgso4干燥并在真空中蒸发以得到作为棕色油的中间体酰胺。步骤4.(4-((2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苄基)氨基甲酸叔丁酯:不经进一步纯化,将来自步骤3的物质((4-(((3-戊烷酰氨基喹啉-4-基)氨基)甲基)苄基)氨基甲酸叔丁酯,222mg,1当量,479μmol)溶解在etoh(20ml)中,并加入在水(200μl)中的氢氧化钠(38.3mg,2当量,958μmol)。将反应混合物回流4-6h并通过uplc监测。反应结束后,将溶剂在减压下除去,将残余物溶解在etoac中,并用水洗涤。将有机层经mgso4干燥,蒸发至干燥。将粗萃取物使用柱色谱法(hex-etoac)纯化以得到作为琥珀色油状物质的期望的化合物(23.7mg,11.1%)。步骤5.1-(4-(((叔丁氧基羰基)氨基)甲基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉5-氧化物:向在dcm/meoh(19:1,3.5ml)中的步骤4的产物((4-((2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苄基)氨基甲酸叔丁酯,31.5mg,1当量,70.9μmol)中加入3-氯过氧苯甲酸(3-chlorobenzoperoxoic acid)(116mg,9.5当量,673μmol),并将反应物在50℃搅拌3h。将溶剂除去并将粗残余物重新溶解在dma中,并通过hplc纯化以产生12.3mg(37.7%)琥珀色油状物质。步骤6.(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苄基)氨基甲酸叔丁酯:向步骤5的产物(12.3mg,1当量,26.7μmol)在ch2cl2(400μl)中的溶液中加入异氰酸苯甲酰酯{1.3当量}并在45℃搅拌3h。(b)将溶剂在真空中蒸发并将残余物溶解在无水meoh(50μl)中,随后加入过量的甲醇钠(350μl),并进一步在80℃搅拌2h。将溶剂在减压下除去,在ch2cl2和水之间分配,将有机层经mgso4干燥并浓缩。将残余物使用hplc纯化以产生10.7mg(87%)作为白色固体的标题化合物。步骤7.1-(4-(氨基甲基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺:向步骤6的产物(10mg,1当量,22μmol)在dcm(273μl)中的溶液中加入20%v/v tfa(1ml)并在室温搅拌2h。通过uplc监测反应,产物峰在2.60min洗脱。将反应混合物在真空中蒸发,将粗萃取物重新溶解在dma中并使用hplc纯化,产生作为白色固体的标题化合物(4.9mg,62.8%)lc-ms:m/z359.48[m+1]+;保留时间=1.99min。1h nmr(400mhz,dmso)δh/ppm 11.57(s,1h),8.14(dd,j=5.5,2.6hz,1h),7.43(s,2h),7.41(d,j=1.9hz,2h),7.39(s,1h),7.37-7.31(m,2h),7.10(dd,j=18.7,8.3hz,2h),5.85(s,1h),3.99(dt,j=11.2,5.7hz,2h),3.43(s,1h),1.72(dt,j=15.3,8.2hz,2h),1.41-1.34(m,2h),0.90-0.85(m,3h)。实施例2. 1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(e104)的制备方案2.试剂:(i)net3,ch2cl2;(ii)zn,hcoonh4,meoh;(iii)c4h9cocl,net3,thf;(iv)naoh,etoh;(v)mcpba,chcl3;(vi)甲苯磺酰氯,ch2cl2;nh4oh(vii)20%tfa,dcm,rt,2h.步骤1.(4-(((3-硝基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯:向4-氯-3-硝基喹啉(1.55g,1当量,7.43mmol)在dcm(15ml)中的溶液中加入(4-(氨基甲基)苯基)氨基甲酸叔丁酯(1.65g,1当量,7.43mmol)和三乙胺(1.13g,1.55ml,1.5当量,11.1mmol)。将混合物在40℃回流1h。通过uplc监测反应进程。在45min内实现反应物向期望产物的完全转化,形成微黄色沉淀物。将反应混合物冷却至室温,过滤并在真空下干燥以得到作为亮黄色固体的期望的化合物(3.35g)。hplc rt=2.86min;m/z=395.3[m+h]。步骤2.(4-(((3-氨基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯:向步骤1的产物((4-(((3-硝基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯,3.35g,1当量,8.49mmol)在meoh(2.2ml)中的悬浮液中加入锌(2.78g,5当量,42.5mmol)和甲酸铵(2.68g,5当量,42.5mmol)。将反应混合物在室温搅拌20min(以产生灰色悬浮液)并通过uplc监测。产物立即开始形成。20分钟以后,将反应混合物穿过硅藻土过滤并将溶剂在真空中蒸发。将残余物溶解在水中,用etoac(3×20ml)萃取,用水洗涤并经mgso4干燥。在真空下除去溶剂以得到3.1g(100%)作为粘性的、蓬松的、深黄色物质的标题化合物。hplc rt=2.14;m/z=365.3[m+h]。步骤3.(4-(((3-戊烷酰氨基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯:向冷却至0℃的在无水etoac(40ml)中的步骤2的粗产物((4-(((3-氨基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯,1500mg,1当量,4.116mmol)中,加入预先冷却的三乙胺(541.4mg,746μl,1.3当量,5.351mmol)。将反应物在室温搅拌15分钟。此后,在-10-0℃逐滴加入在etoac(20ml)中的戊酰氯(545.9mg,537.3μl,1.1当量,4.527mmol),并将反应混合物进一步搅拌30min,并通过uplc监测。将反应混合物用水洗涤,将有机级分经mgso4干燥并在减压下蒸发以得到1.85g作为棕色油的标题化合物,其在真空下干燥以后变成蓬松的棕色固体。lcms rt=2.30min;m/z=449.4[m+h]。步骤4.(4-((2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸叔丁酯:将步骤3的粗产物((4-(((3-戊烷酰氨基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯,1.846g,1当量,4.115mmol)溶解在etoh(26ml)中并用在h2o(4ml)中的氢氧化钠(329.2mg,2当量,8.231mmol)处理。将反应混合物在80℃回流5h并通过uplc监测进程。结束后,将溶剂在减压下除去并将残余物溶解在etoac中,并用水洗涤。将有机层经mgso4干燥并蒸发至干燥。加入饱和nahco3溶液并将产物用etoac萃取,经mgso4干燥并在真空中干燥。将干燥的棕色油状粗萃取物(1700mg)不经进一步纯化用于下一步。lcms rt=2.63;m/z=431.3[m+h]。步骤5.1-(4-((叔丁氧基羰基)氨基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉5-氧化物:不经进一步纯化,将在ch2cl2/meoh(19:1,14ml)中的(4-((2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸叔丁酯(1700mg,1当量,3.948mmol)}用3-氯过氧苯甲酸(2.725g,4当量,15.79mmol)处理并在50℃搅拌3h。将反应混合物在减压下蒸发,用etoac萃取,接连地用饱和nahco3溶液和水洗涤。将有机级分经mgso4干燥并将溶剂在减压下蒸发以得到粗制的产物(1.8g,收率100%)。将20mg粗产物溶解在dma(500μl)中并在hplc上纯化进行表征。将剩余物质不经进一步纯化用于下一步。lcms rt=3.08;m/z=447.3[m+h]。步骤6.(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸叔丁酯:在0-10℃向粗制的(4-((2-丁基-5-(l1-氧烷基)-1h-5l4-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸叔丁酯(1742mg,1当量,3.901mmol)在ch2cl2(125ml)中的溶液中逐滴加入4-甲基苯磺酰氯(966.8mg,1.3当量,5.071mmol),随后加入28-38%氢氧化铵(125.8g,0.14l,920当量,3.589mol)。将混合物在室温搅拌2h并通过uplc监测。结束后,将反应物用水稀释并将有机级分分离,并用2m hcl洗涤,经mgso4干燥并在真空中除去溶剂以得到506mg粗产物,将其不经进一步纯化用于下一步。lcms rt=2.66min;m/z=446.3[m+h]。步骤7.1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺:向(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸叔丁酯的粗萃取物(506mg,1当量,49μmol)在ch2cl2(9ml)中的溶液中加入tfa(2.3ml)。将混合物在室温搅拌1h并在真空中除去溶剂。将残余物的一部分通过hplc纯化以产生标题化合物,而将剩余的粗残余物直接用在后续步骤中。lc-ms:m/z 345.4[m+1]+;保留时间=2.39min。1h nmr(400mhz,dmso)δh/ppm 7.98(dd,j=8.3,1.3hz,2h),7.79(dd,j=8.4,1.3hz,2h),7.39(d,j=1.3hz,2h),7.36(s,2h),6.94(dd,j=26.7,8.6hz,2h),5.87(s,1h),2.99-2.94(m,2h),2.86(d,j=63.6hz,2h),2.02(d,j=51.5hz,2h),0.87(t,j=7.3hz,3h)。实施例3.(4-氨基-1-(4-氨基苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲醇[e136]的制备方案3.试剂:(i)acoch2cocl,net3,thf;(ii)et3n,etoh;(v)mcpba,chcl3:(vi)异氰酸苯甲酰酯,ch2cl2;naoh,meoh(vii)20%tfa,dcm,rt,2h.步骤1.乙酸2-((4-((4-((叔丁氧基羰基)氨基)苄基)氨基)喹啉-3-基)氨基)-2-氧代乙酯:向冷却至-10-0℃的在etoac(15ml)中的(4-(((3-氨基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯(实施例2,步骤2)(2.0g,1当量,5.28mmol)中加入预先冷却的tea三乙胺(2.21ml,3当量,15.9mmol)。此后,在-10-0℃逐滴加入在etoac(7.5ml)中的乙酸2-氯-2-氧代乙酯(938mg,1.5当量),并将反应混合物进一步搅拌30min。通过uplc监测反应。将反应混合物用水洗涤并将有机级分经mgso4干燥并在真空中蒸发以得到作为棕色油的标题化合物,其变为蓬松的棕色固体,将其直接用在下一步中。hplc rt=1.93mg;m/z=479.3[m+h]。[应当指出,由于碱性反应条件,产物被某种环化产物污染。]步骤2.乙酸(1-(4-((叔丁氧基羰基)氨基)苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲酯:将前一步的粗产物(约5.3mmol)溶解在etoh(15ml)中并用tea(4.956g,6.83ml,7当量,48.98mmol)处理。将混合物加热至80℃保持2h。用uplc监测反应直到完全转化。将溶剂在减压下蒸发以产生1500mg粗产物。hplc rt=2.96min;m/z=447.3[m+h]。步骤3.2-(乙酰氧基甲基)-1-(4-((叔丁氧基羰基)氨基)苄基)-1h-咪唑并[4,5-c]喹啉5-氧化物:不经进一步纯化,将在ch2cl2/meoh(19:1,15ml)中的乙酸2-((4-((4-((叔丁氧基羰基)氨基)苄基)氨基)喹啉-3-基)氨基)-2-氧代乙酯(1500.00mg,1当量,3.2291mmol)用3-氯过氧苯甲酸(1.6717g,3当量,9.6874mmol)处理并在50℃搅拌3h。将反应混合物在减压下蒸发,用etoac萃取,用水洗涤。将有机级分经mgso4干燥并将溶剂在减压下蒸发以得到500mg粗产物。hplc rt=3.14min;m/z=463.2[m+h]。步骤6.(4-((4-氨基-2-(羟甲基)-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸叔丁酯:将来自以上反应的粗制物质(500.00mg,1当量,1.0811mmol)在ch2cl2(13ml)中的溶液用异氰酸苯甲酰酯(209.61mg,1.3当量,1.4054mmol)处理并在45℃搅拌3h。将溶剂在真空中蒸发并将残余物溶解在无水meoh(5ml)中,随后加入过量的甲醇钠(3ml)。在80℃搅拌2h以后,将溶剂在减压下除去,将残余物在ch2cl2和水之间分配,并将有机层经mgso4干燥并浓缩。将残余物不经进一步纯化用于下一步。hplc rt=2.64;m/z=420.4。步骤7.(4-氨基-1-(4-氨基苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲醇:在含有搅拌棒的圆底烧瓶中向粗制的(4-((4-氨基-2-(羟甲基)-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苄基)氨基甲酸叔丁酯(50mg,1当量)的溶液中加入tfa(1ml)并将反应物在环境温度搅拌1h。将溶剂在减压下蒸发以提供粗残余物,将其通过hplc纯化以产生作为黄色油的期望产物。hplc rt=1.04;m/z=320.2[m+h]。实施例4.(4-氨基-1-(4-(氨基甲基)苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲醇(e75)的制备方案4.试剂:(i)acoch2cocl,net3,thf;(ii)et3n,etoh;(v)mcpba,chcl3;(vi)异氰酸苯甲酰酯,ch2cl2;naoh,meoh(vii)20%tfa,dcm,rt,2h.步骤1.乙酸2-((4-((4-(((叔丁氧基羰基)氨基)甲基)苄基)氨基)喹啉-3-基)氨基)-2-氧代乙酯:将(4-(((3-氨基喹啉-4-基)氨基)甲基)苄基)氨基甲酸叔丁酯(实施例1,步骤2)(2.00g,1当量,5.28mmol)溶解在etoac(15ml)中并冷却至-10-0℃。加入预先冷却的三乙胺(1.60g,2.21ml,3当量,15.9mmol)并将反应物搅拌15分钟,在此时在-10-0℃逐滴加入在etoac(7.5ml)中的乙酸2-氯-2-氧代乙酯(938mg,739μl,1.3当量,6.87mmol)。将反应混合物搅拌30min并通过uplc监测。将有机溶液用水洗涤并将有机级分经mgso4干燥并在真空中蒸发以得到作为棕色油的标题产物(2.45g),其在真空下干燥以后变成蓬松的棕色固体。hplc rt=1.93;m/z=479.3[m+h]。步骤2.乙酸2-((4-((4-(((叔丁氧基羰基)氨基)甲基)苄基)氨基)喹啉-3-基)氨基)-2-氧代乙酯:将来自前一步的粗产物(2.450g,1当量,5.120mmol)溶解在etoh(15ml)中并用三乙胺(3.626g,5.00ml,7当量,35.84mmol)处理。将反应物加热至80℃保持12h并用uplc监测。将反应物在1mhcl和etoac之间分配。将有机级分经mgso4干燥并在减压下蒸发以提供粗产物,将其不经纯化用于后续步骤。hplc rt=2.42;m/z=461.3。步骤3.2-(乙酰氧基甲基)-1-(4-(((叔丁氧基羰基)氨基)甲基)苄基)-1h-咪唑并[4,5-c]喹啉5-氧化物:将在ch2cl2/meoh(19:1,12ml)中的前一步的产物(1000.00mg,1当量,2.1714mmol)用3-氯过氧苯甲酸(1.1241g,3当量,6.5142mmol)处理并在50℃搅拌3h。将反应混合物在减压下蒸发,用etoac萃取,用水洗涤。将有机级分经mgso4干燥并将溶剂在减压下蒸发以得到粗产物,将其不经进一步纯化用在下一步中。hplc rt=3.06;m/z=477.3。步骤4.(4-((4-氨基-2-(羟甲基)-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苄基)氨基甲酸叔丁酯:将前一个反应的粗产物(500.00mg,1当量,1.0493mmol)溶解在ch2cl2(13ml)中并用异氰酸苯甲酰酯(238.96mg,1.3当量,1.36mmol)处理并在45℃搅拌3h。将溶剂在真空中蒸发并将残余物溶解在无水meoh(5ml)中,随后加入甲醇钠(1当量),并进一步在80℃搅拌2h。将溶剂在减压下除去,在ch2cl2和水之间分配,将有机层经mgso4干燥并在真空中浓缩。lcms显示在2.53min和2.72min的两个主要峰,其分别对应于乙酰化的和脱乙酰化的产物。将残余物不经进一步纯化用于下一步。hplc rt=2.53;m/z=434.4[m+h]。步骤5.(4-氨基-1-(4-(氨基甲基)苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲醇:向来自前一步的粗制产物(450.0mg,1当量,946.3μmol)的溶液中加入20% tfa(10ml)并将反应物在环境温度搅拌1h。将溶剂在减压下蒸发以提供固体。将粗制物在hplc上纯化,产生45mg标题化合物。hplc=0.32mg;m/z=334[m+h]。实施例5.从咪喹莫特衍生出的连接基-有效负载.lp#1:6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-n-(1-(1-(1-异丁基-1h-吡咯并[3,2-c]喹啉-4-基氨基)-1-氧代-5-脲基戊烷-2-基氨基)-3-甲基-1-氧代丁烷-2-基)己酰胺[mcvalcit-咪喹莫特]的制备将在dmf(2ml)中的咪喹莫特(10mg,42μmol)和2,6-二甲基吡啶(15μl,0.13mmol)一起在室温搅拌15min,然后加入hobt(8.3mg,54μmol)、hatu(19mg,50μmol)和mcvalcit-oh(21mg,46μmol)。将反应混合物进一步在室温搅拌12h。将粗制的反应混合物使用(10%至95% acn在水中的溶液,含有0.05% tfa)通过反相制备型hplc纯化。将含有期望的化合物的级分在高真空下干燥以产生12.9mg(44.5%)作为黄色油状物质的标题化合物。lc-ms(方案a):m/z 690.4[m+1]+;保留时间=2.32min。1h nmr(400mhz,dmso)δh/ppm 1h nmr(400mhz,dmso)δ8.79(s,1h),8.51(s,1h),8.49(d,j=6.5hz,1h),8.43(d,j=8.4hz,2h),7.86(ddd,j=14.1,12.0,5.7hz,4h),6.99(s,2h),4.89(s,1h),4.61(d,j=7.5hz,2h),4.50(d,j=7.5hz,1h),4.29-4.22(m,1h),3.37(t,j=7.0hz,3h),3.04(dd,j=10.9,6.8hz,2h),2.94(s,1h),2.23-2.08(m,5h),1.53-1.40(m,10h),1.18(dd,j=13.8,9.4hz,4h),0.95(t,j=6.5hz,9h),0.90(d,j=6.8hz,4h),0.87-0.81(m,8h)。lp#2:6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-n-(1-异丁基-1h-吡咯并[3,2-c]喹啉-4-基)己酰胺[mc-咪喹莫特]向6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸(15.0mg,71.0μmol)在dma(400μl)中的溶液中加入hobt(15.2mg,99.4μmol)、hatu(32.4mg,85.2μmol)和2,6-二甲基吡啶(24.7μl,213μmol)。在室温搅拌15min以后,逐滴加入咪喹莫特(18.7mg,78.1μmol)和2,6-二甲基吡啶(24.7μl,213μmol)在dma(600μl)中的溶液,并将反应混合物在室温搅拌72h。将粗制混合物在制备型hplc上纯化以得到e131(4.1mg,25%),lc-ms(方案a):m/z 764.5[m+1]+;保留时间=2.78min。1h nmr(400mhz,dmso)δh/ppm 8.78(s,1h),8.44(d,j=9.9hz,2h),7.92(t,j=7.3hz,1h),7.85(t,j=7.7hz,1h),7.01(s,2h),4.62(d,j=7.5hz,3h),3.43(t,j=7.1hz,2h),2.79(t,j=7.3hz,2h),2.21(dt,j=14.9,7.2hz,1h),1.76-1.66(m,2h),1.56(dt,j=14.9,7.3hz,2h),1.40-1.31(m,2h),0.97(d,j=6.6hz,6h),0.94(s,1h)。实施例6.从瑞喹莫德(r848)衍生出的连接基-有效负载lp#3.4.6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-n-(1-(1-(2-(乙氧基甲基)-1-(2-羟基-2-甲基丙基)-1h-吡咯并[3,2-c]喹啉-4-基氨基)-1-氧代-5-脲基戊烷-2-基氨基)-3-甲基-1-氧代丁烷-2-基)己酰胺[mcvalcit-瑞喹莫德]将dma(0.5ml)加入含有瑞喹莫德(15mg,48μmol)和hobt(9.5mg,62μmol)的管形瓶中。用注射器加入2,6-二甲基吡啶(17μl,0.14mmol)。将反应混合物在室温搅拌12h,并将粗产物使用(10%乙腈/90%h2o持续5分钟,然后用10%乙腈至95%乙腈在h2o中的溶液历时10分钟,各溶剂含有0.05% tfa)通过反相制备型hplc纯化,产生15.8mg(42.7%)作为微黄色油的标题化合物。lc-ms(方案a):m/z 764.5[m+1]+;保留时间=2.33min。1h nmr(400mhz,dmso)δh/ppm 8.76(d,j=8.4hz,1h),8.62-8.44(m,1h),8.40(d,j=8.4hz,1h),7.82(ddd,j=22.6,15.6,7.9hz,4h),7.00(s,2h),4.95(s,1h),4.39-4.10(m,2h),3.58(dq,j=14.0,7.0hz,3h),3.41-3.28(m,3h),3.11-3.01(m,2h),2.96(d,j=4.6hz,1h),2.22-2.10(m,3h),1.49(dt,j=14.4,7.2hz,7h),1.18(dd,j=14.2,7.3hz,13h),0.91(d,j=6.7hz,3h),0.84(dt,j=6.8,5.6hz,8h)。lp#4:2-(乙氧基甲基)-1-(2-羟基-2-甲基丙基)-1h-咪唑并[4,5-c]喹啉-4-基氨基甲酸4-(2-(2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯[mcvalcitpabc-瑞喹莫德]使用玻璃注射器向瑞喹莫德(20mg,64μmol)在dmf(300μl)中的溶液中加入2,6-二甲基吡啶(22μl,0.19mmol)。然后将反应物在室温搅拌15min。此后,使用玻璃注射器加入hobt(12mg,76μmol)和mcvalcit-pab-pnp(40mg,70μmol)在dmf(300μl)中的溶液,并将反应混合物在室温搅拌过夜。加入水(5ml)并将产物用etoac(3x 5ml)萃取。将有机层经无水mgso4干燥。将有机级分在减压下蒸发,然后将粗萃取物溶解在dma(500μl)中并使用(10%乙腈/90% h2o持续5分钟,然后10%乙腈至95%乙腈在h2o中的溶液历时10分钟,各溶剂含有0.05% tfa)通过反相制备型hplc纯化以产生6.5mg(11.2%)作为白色固体的标题化合物。lc-ms(方案a):m/z 913.5[m+1]+;保留时间=2.88min。1h nmr(400mhz,dmso)δh/ppm10.02(s,1h),8.65(d,j=8.4hz,1h),8.18-8.02(m,2h),7.80(d,j=8.7hz,2h),7.65(d,j=8.6hz,2h),7.44(d,j=8.6hz,1h),7.00(s,2h),5.25(s,1h),4.39(dd,j=16.2,9.1hz,2h),4.24-4.13(m,1h),3.56(q,j=7.0hz,2h),3.37(t,j=7.0hz,2h),2.95(s,4h),2.79(s,5h),2.20-2.06(m,3h),1.96(s,6h),1.48(dt,j=15.0,7.3hz,6h),1.18(dt,j=14.0,7.6hz,10h),0.84(dd,j=12.2,6.8hz,7h)。lp#5:6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-n-(2-(乙氧基甲基)-1-(2-羟基-2-甲基丙基)-1h-咪唑并[4,5-c]喹啉-4-基)己酰胺[mc-瑞喹莫德]向6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸(13.0mg,61.5μmol)在dma(400μl)中的溶液中加入2,6-二甲基吡啶(21.4μl,185μmol)和hobt(13.2mg,86.2μmol)。将该混合物在室温搅拌15min并用瑞喹莫德(21.3mg,67.7μmol)和2,6-二甲基吡啶(21.4μl,185μmol)在dma(300μl)中的溶液处理。在室温72h以后,将粗制混合物在制备型hplc上纯化,产生8.6mg(27.6%)作为白色固体的e130,lc-ms(方案a):m/z764.5[m+1]+;保留时间=2.80min。1h nmr(400mhz,dmso)δh/ppm8.74(d,j=8.3hz,1h),8.37(d,j=8.4hz,1h),7.86(t,j=7.5hz,1h),7.76(t,j=7.7hz,1h),7.01(s,2h),4.85-4.79(m,1h),3.58(dd,j=14.0,7.0hz,6h),3.42(t,j=7.0hz,3h),2.80(t,j=7.3hz,2h),1.75-1.66(m,2h),1.56(dt,j=14.8,7.3hz,3h),1.39-1.29(m,3h),1.21(s,1h),1.16(t,j=7.0hz,5h)。实施例7.从1-(4-(氨基甲基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(e66)衍生出的连接基-有效负载lp#6:4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苄基氨基甲酸4-(2-(2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯[mcvalcitpabc-e66]将2,6-二甲基吡啶(7.9μl,68μmol)加入1-(4-(氨基甲基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(实施例1)(8.2mg,23μmol)在dma(400μl)中的溶液中并在25℃搅拌15min。hobt(4.2mg,1.2当量,27μmol)。加入在dma(200μl)中的mcvalcitpab-pnp(14mg,25μmol),并将反应混合物在室温搅拌过夜。加入饱和nahco3溶液(3ml)并将产物用ch2cl2(3x5ml)萃取。将有机级分经无水mgso4干燥,并在真空中蒸发溶剂。将粗萃取物溶解在dma(500μl)中并使用(10%乙腈/90% h2o持续5分钟,然后10%乙腈至95%乙腈在h2o中的溶液历时10分钟,各溶剂含有0.05% tfa)通过反相制备型hplc纯化以产生白色固体(16.4mg,74.6%)。lc-ms(方案a):m/z 958.5[m+1]+;保留时间=2.55min。1h nmr(400mhz,dmso)δh/ppm 10.06(s,1h),9.92(dd,j=76.6,35.4hz,2h),8.07(dd,j=14.4,7.5hz,3h),8.02(d,j=8.4hz,1h),7.81(d,j=8.6hz,4h),7.61(d,j=8.5hz,5h),7.58-7.50(m,4h),7.45-7.35(m,5h),7.32(d,j=8.5hz,2h),7.23(d,j=8.5hz,1h),7.00(s,4h),6.32-5.59(m,22h),5.53(s,2h),5.06(d,j=10.3hz,2h),4.43(s,1h),4.42-4.33(m,3h),4.19(t,j=7.7hz,3h),3.37(t,j=7.0hz,6h),3.08-2.90(m,7h),2.25-2.06(m,7h),2.06-1.86(m,3h),1.71(dt,j=14.4,7.2hz,4h),1.59(dd,j=13.5,4.3hz,3h),1.48(dt,j=14.7,7.3hz,14h),1.37(dd,j=14.9,7.4hz,5h),1.24-1.13(m,6h),0.84(dd,j=12.4,6.7hz,19h)。lp#7:n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苄基)-6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰胺.[mc-e66]将e66[1-(4-(氨基甲基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(4.40mg,12.2μmol)]、6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸(3.10mg,14.7μmol)、hatu(6.05mg,1.3当量,15.9μmol)、1h-苯并[d][1,2,3]三唑-1-醇水合物(2.62mg,1.4当量,17.1μmol)和2,6-二甲基吡啶(3.93mg,4.25μl,3当量,36.7μmol)的溶液在室温搅拌1.5小时。将粗制混合物在hplc上纯化以得到期望产物(1.6mg,36.4%)。lc-ms(方案a):m/z552.68[m+1]+;保留时间=3.24min。1h nmr(400mhz,dmso)δh/ppm13.65(s,1h),8.52(dd,j=11.5,7.9hz,1h),7.79(d,j=8.2hz,1h),7.69(dd,j=7.4,3.5hz,2h),7.51(dd,j=14.9,7.0hz,1h),7.01(dd,j=6.3,3.1hz,1h),6.99(s,2h),6.95(s,1h),3.72(dd,j=16.4,8.7hz,1h),3.50(dd,j=14.2,7.1hz,3h),3.41-3.30(m,3h),2.94(s,1h),2.78(s,1h),2.68(s,2h),2.54(s,1h),2.35(t,j=7.4hz,2h),1.95(s,1h),1.56-1.39(m,4h),1.29-1.18(m,2h),1.13(t,j=7.1hz,3h),1.02(t,j=7.1hz,1h)。lp#8:n-(1-(1-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苄基氨基)-1-氧代-5-脲基戊烷-2-基氨基)-3-甲基-1-氧代丁烷-2-基)-6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰胺[mcvalcit-e66]。步骤1.将e66[1-(4-(氨基甲基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺,10mg,1当量,28μmol]、boc-valcit-oh(12mg,1.2当量,33μmol)、hatu(14mg,1.3当量,36μmol)、2,6二甲基吡啶(8.6mg,3当量)和1h-苯并[d][1,2,3]三唑-1-醇水合物(6.0mg,1.4当量,39μmol)在dma(500μl)中的溶液在室温搅拌1h。通过lcms监测反应。将粗制混合物在hplc上纯化以产生1.3mg(m/z=716.9)作为白色固体的产物。将产物用1ml的20% tfa在dcm中的溶液处理1h。将溶剂蒸发并将物质不经纯化用在下一步中。步骤2.向步骤1的产物(1.10mg,1当量,1.79μmol)在dma(300μl)中的溶液中加入6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸(453μg,1.2当量,2.14μmol)、2-(3h-[1,2,3]三唑并[4,5-b]吡啶-3-基)-1,1,3,3-四甲基异脲鎓六氟磷酸盐(v)(883μg,1.3当量,2.32μmol),并将反应混合物在室温搅拌1.5h。将粗制混合物在制备型hplc上纯化以得到期望产物,lc-ms(方案a):m/z 808.99[m+1]+;保留时间=2.90min。1h nmr(400mhz,dmso)δh/ppm 8.31(dd,j=7.3,4.6hz,1h),7.95(d,j=7.7hz,1h),7.90(d,j=7.9hz,1h),7.80(d,j=7.5hz,1h),7.77-7.71(m,1h),7.65-7.59(m,1h),7.40(d,j=2.1hz,1h),7.36(d,j=8.7hz,1h),7.21-7.16(m,2h),7.00(s,2h),5.93(s,1h),5.79(d,j=1.8hz,2h),3.31(d,j=3.2hz,2h),2.67(dt,j=3.9,1.9hz,7h),2.52(d,j=1.7hz,6h),2.34-2.31(m,7h),1.52-1.43(m,3h),1.37(dt,j=16.4,7.3hz,3h),1.17(d,j=7.5hz,1h),0.87(t,j=7.3hz,7h),0.82(d,j=2.4hz,2h),0.74(d,j=6.7hz,6h)。实施例8.从1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(e104)衍生出的连接基-有效负载lp#9.4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基氨基甲酸4-(2-(2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯[mcvalcitpabc-e104]向1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(e104,实施例2)(9.1mg,26μmol)在dma(500μl)中的溶液中加入mcvalcit-pab-pnp(23mg,32μmol)、2,6-二甲基吡啶(9.2μl,79μmol)和hobt(5.6mg,37μmol)。将混合物在室温搅拌过夜。将粗制混合物通过制备型hplc纯化以产生琥珀色粘性固体(36.2mg,60.3%)。lc-ms(方案a):m/z944.1[m+1]+;保留时间=3.27min。1h nmr(400mhz,dmso)δh/ppm 9.97(s,1h),7.79(d,j=1.2hz,1h),7.59(d,j=8.8hz,1h),7.40(d,j=9.2hz,1h),7.32(d,j=8.7hz,1h),7.00(s,1h),6.99(s,2h),6.97(d,j=6.6hz,1h),6.51(s,2h),5.39(s,1h),5.04(s,1h),2.77(d,j=2.7hz,2h),2.69-2.65(m,11h),2.33(dt,j=3.9,2.1hz,9h),2.08(s,3h),1.95(s,4h),1.24(s,3h),0.89-0.86(m,2h),0.85(t,j=1.8hz,2h),0.84-0.80(m,2h)。lp#10:n-(1-(1-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基氨基)-1-氧代-5-脲基戊烷-2-基氨基)-3-甲基-1-氧代丁烷-2-基)-6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰胺[mcvalcit-e104]。步骤1.将e104[1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺,9.20mg,1当量,26.6μmol]在500μl dma中的溶液用boc-valcit-oh(13.0mg,1.3当量,34.6μmol)、1h-苯并[d][1,2,3]三唑-1-醇水合物(6.12mg,1.5当量,39.9μmol)、hatu(14.2mg,1.4当量)和2,6-二甲基吡啶(8.5mg,3当量)处理。将反应物在室温搅拌1h并通过lcms监测。在1h以后注意到完全转化。将粗制物质通过hplc纯化以产生4.2mg(22.5%)作为白色固体的期望产物。hplc rt=3.03;m/z=702.5[m+h]。步骤2.将步骤1的产物用1ml的20% tfa在dcm中的溶液处理。1h以后,lcms指示反应结束。将溶剂蒸发以产生粗产物,将其立即用在dma(300μl)中的6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸(1.3mg,1.2当量,6.4μmol)、hatu(2.6mg,1.3当量,6.9μmol)、1h-苯并[d][1,2,3]三唑-1-醇水合物(1.1mg,1.4当量,7.4μmol)和2,6-二甲基吡啶(1.7mg,1.8μl,3当量,16μmol)处理。在室温搅拌1h以后,将产物混合物通过hplc纯化,产生1.1mg标题化合物。hplc rt=2.97;m/z 795.5[m+h]。lp#11:n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)-6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰胺[mc-e104]将1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(实施例2)(10.0mg,28.95μmol)]、6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸(7.337mg,34.74μmol)、hatu(14.31mg,37.63μmol)、1h-苯并[d][1,2,3]三唑-1-醇水合物(6.206mg,40.53μmol)和2,6-二甲基吡啶(9.3mg,10.1μl,86.8μmol)的溶液在室温搅拌1.5小时。将粗制混合物在hplc上纯化以得到作为淡黄色固体的期望产物(11.2mg,71.8%)。lc-ms(方案a):m/z 538.6[m+1]+;保留时间=3.47min。1h nmr(400mhz,dmso)δh/ppm 9.86(s,1h),7.96(d,j=8.1hz,1h),7.79(d,j=8.2hz,1h),7.65-7.60(m,1h),7.52(dd,j=9.0,2.9hz,3h),7.40-7.35(m,1h),7.01(d,j=4.0hz,2h),7.00-6.98(m,2h),6.97(s,2h),6.96(d,j=2.3hz,1h),5.89(s,1h),3.37(dd,j=8.8,5.3hz,3h),2.98(d,j=7.5hz,1h),2.95(d,j=3.2hz,1h),2.24(dd,j=12.8,5.4hz,2h),1.72(ddd,j=12.2,7.4,3.6hz,2h),1.58-1.46(m,3h),1.38(dt,j=14.5,7.5hz,2h),1.27-1.17(m,2h),0.85(t,j=3.7hz,2h)。实施例9.从嘎德莫特衍生出的连接基-有效负载.lp#12:(4-氨基-1-(2-羟基-2-甲基丙基)-1h-咪唑并[4,5-c]喹啉-2-基)甲基(乙基)氨基甲酸4-(2-(2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯[mcvalcitpabc-嘎德莫特]向1-(4-氨基-2-((乙基氨基)甲基)-1h-咪唑并[4,5-c]喹啉-1-基)-2-甲基丙烷-2-醇双(2,2,2-三氟乙酸酯)(10mg,18μmol)在dma(500μl)中的溶液中加入2,6-二甲基吡啶(13μl,0.11mmol)并在室温搅拌10min。然后,加入1h-苯并[d][1,2,3]三唑-1-醇水合物(4.0mg,26μmol)和mcvalcitpab-pnp(16mg,22μmol),并将反应物进一步在室温搅拌12h。将粗制混合物在制备型hplc上纯化以产生琥珀色粘性固体(7.4mg,43.5%)。lc-ms(方案a):m/z 958.5[m+1]+;保留时间=2.74min。1hnmr(400mhz,dmso)δh/ppm 9.94(d,j=34.6hz,1h),8.88(s,2h),8.45(d,j=57.6hz,1h),8.11(d,j=9.2hz,1h),8.06(d,j=7.5hz,1h),7.79(dd,j=8.4,3.6hz,2h),7.68(t,j=7.7hz,1h),7.51(t,j=7.7hz,1h),7.43(s,1h),7.31(s,1h),7.16(s,1h),6.99(s,2h),6.93(d,j=9.2hz,1h),6.04(s,1h),5.01(d,j=38.4hz,2h),4.38(dd,j=13.4,8.1hz,3h),4.23-4.14(m,10h),3.36(t,j=7.1hz,3h),3.06-2.91(m,2h),2.22-2.07(m,2h),2.01-1.91(m,1h),1.52-1.42(m,5h),1.18(dt,j=15.4,7.8hz,5h),1.08(s,7h),0.84(dd,j=11.7,6.8hz,7h)。lp#13:n-((4-氨基-1-(2-羟基-2-甲基丙基)-1h-咪唑并[4,5-c]喹啉-2-基)甲基)-6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-n-乙基己酰胺[mc-嘎德莫特]向6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸(4.0mg,19μmol)在dma(400μl)中的溶液中加入hatu(8.6mg,23μmol)、hobt(4.1mg,27μmol)和2,6-二甲基吡啶(6.6μl,57μmol)。在室温搅拌15min以后,逐滴加入三氟乙酸嘎德莫特(10mg,19μmol)和2,6-二甲基吡啶(6.1mg,6.6μl,3当量,57μmol)在dma(600μl)中的溶液,并将反应混合物在室温搅拌12h。将粗制混合物在制备型hplc上纯化以得到6.6mg(68.8%)白色固体,lc-ms(方案a):m/z506.6[m+1]+;保留时间=2.47min。1h nmr(400mhz,dmso)δh/ppm 13.65(s,1h),8.52(dd,j=11.5,7.9hz,1h),7.80(t,j=8.2hz,1h),7.71-7.65(m,2h),7.51(dd,j=15.0,7.0hz,1h),7.01(dd,j=6.2,3.0hz,1h),6.99(s,2h),3.72(dd,j=16.4,8.7hz,1h),3.50(dd,j=14.2,7.2hz,2h),3.40-3.30(m,2h),2.86(d,j=64.1hz,1h),2.71-2.64(m,3h),2.54(s,1h),2.35(t,j=7.4hz,1h),1.50(ddd,j=12.2,11.4,6.1hz,2h),1.24(tdd,j=16.0,10.1,6.2hz,2h),1.13(t,j=7.1hz,2h),1.02(t,j=7.1hz,1h)。实施例10.从4-氨基-1-(4-(氨基甲基)苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲醇(e75)衍生出的连接基-有效负载lp#14:4-((4-氨基-2-(羟甲基)-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苄基氨基甲酸4-(2-(2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯[mcvalcitpabc-e75]向(4-氨基-1-(4-(氨基甲基)苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲醇(实施例4)(17mg,51μmol)在dma(500μl)中的溶液中加入2,6-二甲基吡啶(7.6mg,71μmol)并将混合物在室温搅拌15min。加入1h-苯并[d][1,2,3]三唑-1-醇水合物(23mg,25μl,0.15mmol)和(4-硝基苯基)碳酸4-(2-(2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯(mcvalcitpab-pnp)(49mg,66μmol),并将混合物在室温搅拌过夜。将标题产物通过hplc纯化以产生白色固体(5.0mg,10%)。lc-ms(方案a):m/z 932.05[m+1]+;保留时间=2.86min。1hnmr(400mhz,dmso)δh/ppm 9.97(s,1h),7.79(dd,j=8.5,4.2hz,2h),7.58(d,j=8.7hz,1h),7.26(d,j=8.7hz,1h),7.20(d,j=8.2hz,2h),7.08(d,j=8.3hz,2h),7.00(s,2h),6.99-6.96(m,1h),6.02(s,1h),4.93(s,1h),4.82(d,j=2.1hz,1h),4.38(dd,j=7.9,2.5hz,1h),4.17(dd,j=23.8,6.5hz,2h),4.00(d,j=53.7hz,1h),3.47(d,j=14.5hz,3h),3.23(d,j=2.3hz,2h),2.87(d,j=64.1hz,2h),2.68(dt,j=4.1,2.1hz,4h),2.33(dt,j=4.0,2.0hz,3h),2.09-2.09(m,2h),1.51(d,j=7.6hz,1h),1.47(dd,j=5.2,2.5hz,2h),1.20(d,j=35.8hz,3h),0.86(d,j=6.8hz,3h),0.83(d,j=6.7hz,3h)。lp#15:n-(4-((4-氨基-2-(羟甲基)-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苄基)-6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰胺[mc-e75]向(4-氨基-1-(4-(氨基甲基)苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲醇(实施例4)(17.00mg,50.99μmol)在dma(500μl)中的溶液中加入6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸(12.92mg,61.19μmol)、hatu(25.20mg,66.29μmol)、1h-苯并[d][1,2,3]三唑-1-醇水合物(10.93mg,71.39μmol)和2,6-二甲基吡啶(17.7μl,153.0μmol)。将反应物在室温搅拌1h。将粗制混合物在hplc上纯化以产生白色固体产物(2.3mg);lc-ms(方案a):m/z526.60[m+1]+;保留时间=2.43min。1h nmr(400mhz,dmso)δh/ppm 13.60(s,1h),8.24(t,j=6.1hz,1h),7.95(d,j=8.3hz,1h),7.79(d,j=8.3hz,1h),7.63(t,j=7.8hz,1h),7.37(t,j=7.7hz,1h),7.18(d,j=8.2hz,2h),7.08(d,j=8.2hz,2h),7.02-7.00(m,1h),6.99(s,3h),6.02(s,1h),4.82(s,2h),4.19(d,j=6.0hz,2h),3.33(t,j=7.0hz,3h),2.51(d,j=3.8hz,1h),2.07(t,j=7.4hz,2h),1.51-1.41(m,4h),1.19-1.11(m,2h)。实施例11.从(4-氨基-1-(4-氨基苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲醇[e136]衍生出的连接基-有效负载lp#16:4-((4-氨基-2-(羟甲基)-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基氨基甲酸4-(2-(2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯[mcvalcitpabc-e136]向粗制的4-氨基-1-(4-氨基苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲醇(20.00mg,62.62μmol)在dma(500μl)中的溶液中加入2,6-二甲基吡啶(8.724mg,81.41μmol)并在室温搅拌15min。然后,加入1h-苯并[d][1,2,3]三唑-1-醇水合物(28.77mg,187.9μmol)和(4-硝基苯基)碳酸4-(2-(2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯(55.44mg,75.15μmol)并在室温搅拌过夜。将粗制混合物在hplc上纯化以产生9.8mg(17%)作为白色固体的产物。lc-ms(方案a):m/z 918.03[m+1]+;保留时间=2.74min。1h nmr(400mhz,dmso)δh/ppm 10.00(d,j=14.4hz,5h),8.07(d,j=2.0hz,3h),8.05(dd,j=4.5,2.7hz,6h),7.85-7.74(m,9h),7.62-7.59(m,11h),7.58(dd,j=4.5,2.4hz,14h),7.43-7.37(m,7h),7.36(t,j=2.5hz,4h),7.34(t,j=2.4hz,5h),7.31(d,j=3.3hz,3h),7.28(d,j=2.2hz,4h),7.03-6.98(m,43h),5.81(s,4h),4.98(s,4h),4.40-4.35(m,3h),4.19(ddd,j=8.7,6.6,4.2hz,8h),2.85(d,j=2.3hz,11h),2.68(dt,j=3.9,1.9hz,8h),2.34(dt,j=6.0,2.0hz,8h),1.50(dd,j=7.6,5.0hz,8h),1.47(d,j=3.4hz,2h),1.45(t,j=3.6hz,3h),1.25-1.13(m,5h),0.86(dt,j=6.7,3.2hz,17h),0.83-0.79(m,11h)。lp#17:n-(4-((4-氨基-2-(羟甲基)-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)-6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰胺[mc-e136]向e136[(4-氨基-1-(4-氨基苄基)-1h-咪唑并[4,5-c]喹啉-2-基)甲醇,50.0mg,1当量,156.6μmol]在dma(500μl)中的溶液中加入6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸(39.7mg,1.2当量,187.9μmol)、hatu(77.39mg,1.3当量,203.5μmol)、1h-苯并[d][1,2,3]三唑-1-醇水合物(33.57mg,1.4当量,219.2μmol)和2,6-二甲基吡啶(50.33mg,54.4μl,3当量,469.7μmol)。将反应物在室温搅拌1h。将粗制混合物在hplc上纯化以产生1.3mg(1.6%)作为白色固体的标题产物。hplc rt=2.87;m/z=513.3[m+h]。实施例11a.准备用于连接连接基和抗体的各种tlr激动剂的制备以本领域技术人员已知的多种方式进行。在方案p1(改编自us 2014/0141033)中概述了一种这样的方法。方案p1:步骤1:在升高的温度(100-120℃)下将4-氯-3-硝基喹啉在二氧杂环己烷中用氨处理以导致化合物1的形成。步骤2-3:在乙醇中钯催化氢化该化合物导致化合物2的形成,其又在己酸中回流以导致2-烷基-1h-咪唑并[4,5-c]喹啉3的形成。步骤4:将该化合物在dmf中用4-硝基苄基溴化物进行选择性烷基化,导致化合物4的形成。用碳酸铯或碳酸钾促进该反应,并可以在室温或升高的温度进行。该转化(在其它咪唑氮处的烷基化)的次要异构体通过硅胶色谱法容易与期望的产物分离。步骤5:在氯仿中用间氯过苯甲酸(mcpba)氧化喹啉,导致n-氧化物5的形成。步骤6:用甲苯磺酰氯和过量的氨处理该中间体,导致氨基喹啉6的形成。步骤7:在升高的温度在乙醇中用铁和氯化铵处理化合物6,导致关键中间体7的形成。步骤8:使用hatu和hobt将boc-保护的5-氨基己酸与化合物7偶联。用tfa去保护,产生化合物8a。通过酰化、磺酰化或氨甲酰化类似地修饰中间体7,以产生化合物8b-8i。将最终的化合物通过硅胶色谱法或制备型hplc纯化。准备用于连接连接基和抗体的各种tlr激动剂的子集的一种替代制备显示在方案p2中。方案p2:该反应序列的起始点是2-丁基-1h-咪唑并[4,5-c]喹啉,如上所述制备。将该化合物在dmf中在室温用4-甲氧基苄基溴化物选择性地烷基化以产生化合物1。用碳酸铯或碳酸钾促进该反应,并可以在室温或升高的温度进行。该转化(在其它咪唑氮处的烷基化)的次要异构体通过硅胶色谱法容易与期望的产物分离。步骤2:在氯仿中用间氯过苯甲酸(mcpba)氧化喹啉,导致n-氧化物2的形成。步骤3:用甲苯磺酰氯和过量的氨处理该中间体,导致氨基喹啉3的形成。步骤4:在氯仿中使用过量的三甲基甲硅烷基碘化物脱甲基化,导致关键苯酚4的形成。在dmf和碳酸铯中用烯丙基氯化物将苯酚烷基化,导致5a的形成。同样地,按照cheng的条件(tetrahedron letters,53,1,第71-75页)将苯酚芳基化,导致5b的形成。简言之,在乙腈中在80℃用碘苯(1.5当量)、cu2o(1mol%)、1h-咪唑-4-甲酸(2mol%)和碳酸铯(2当量)处理中间体4。将期望的产物(5b)使用etoac/hex梯度通过硅胶色谱法纯化。实施例12.adc的制备将在实施例5-11中描述的连接基-有效负载与各种抗体缀合,产生表1中所示的adc。使用下述方法完成所述缀合:方法a:将在pbs中的2mg抗体用12当量的5mm三(2-羧基乙基)膦(tcep)处理,达到约5mg/ml的最终蛋白浓度。将反应物在37℃加热1小时。将在dma中的25当量的连接基-有效负载与足够的dma和pbs一起加入反应物中,以产生约5%(体积/体积)的最终有机物和26.7um(4mg/ml)的最终抗体浓度。将反应物在室温静置90分钟。然后将反应物使用sephadex柱根据生产商的方案缓冲液交换进100% pbs中。使用tcep还原一个小等分试样,并使用hplc-ms试验其载荷,并基于相对峰高计算药物与抗体比率(dar)。使用nanodrop使用蛋白a280igg方法确定adc的浓度,并使用尺寸排阻色谱法分析未还原的等分试样的聚集。将最终的adc在储存前进行过滤灭菌。对于表现出低于6的dar的adc,将粗制的adc使用30kd离心旋转装置离心沉降以浓缩样品,并按照与上述相同的顺序对adc重新进行tcep和连接基-有效负载处理。方法b:将2mg抗体用12当量的5mm三(2-羧基乙基)膦(tcep)处理,并将0.1m pbsph 7.4加入反应物中以具有26.7um(4mg/ml)的最终抗体浓度,并在37℃加热2小时。将反应物使用sephadex柱根据生产商的方案进行缓冲液交换,并使用离心旋转装置用30k过滤器浓缩。将在dma中的25当量的连接基-有效负载与足够的dma和pbs一起加入反应物中,以产生约5%(体积/体积)的最终有机物和26.7um(4mg/ml)的最终抗体浓度。将反应物在室温静置90分钟。然后将反应物使用sephadex柱根据生产商的方案缓冲液交换进100% pbs中。使用tcep还原一个小等分试样,并使用hplc-ms试验其载荷,并基于相对峰高计算药物与抗体比率(dar)。使用nanodrop使用蛋白a280 igg方法确定adc的浓度,并使用尺寸排阻色谱法分析未还原的等分试样的聚集。将最终的adc在储存前进行过滤灭菌。对于表现出低于6的dar的adc,将粗制的adc使用30kd离心旋转装置离心沉降以浓缩样品,并按照与上述相同的顺序对adc重新进行tcep和连接基-有效负载处理。方法c:将2mg抗体用12当量的5mm三(2-羧基乙基)膦(tcep)处理,并将含有5mmedta的0.1m pbs ph 7.4加入反应物中以具有26.7um的最终抗体浓度,并在37℃加热2小时。将反应物使用sephadex柱根据生产商的方案进行缓冲液交换,并使用离心旋转装置用30k过滤器浓缩。将在dma中的25当量的连接基-有效负载与含有5mm edta的0.1m pbs ph7.4(含有5% dma(v/v))一起加入浓缩的还原的抗体中,以产生26.7um的最终抗体浓度(4mg/ml)。将反应物在室温静置1.5小时。然后,将反应物使用sephadex柱根据生产商的方案进行缓冲液交换。使用tcep还原一个等分试样并使用hplc-ms试验其载荷,并计算药物与抗体比率(dar)。使用nanodrop使用蛋白a280 igg方法确定adc的浓度,并使用在uplc上的尺寸排阻色谱法分析未还原的等分试样的聚集。将最终的adc通过过滤灭菌进行纯化。对于表现出低于6的dar的adc,将粗制的adc使用30kd离心旋转装置离心沉降以浓缩样品,并按照与上述相同的顺序对adc重新进行tcep和连接基-有效负载处理。表1:adc的列表实施例13:使用24h或72h测定评价针对ramos-blue细胞的有效负载。对于每种有效负载,在10% dmso在pbs中的溶液中进行5倍系列稀释,以具有1000um至64nm的最终浓度范围。将ramos-blue细胞(invivogen,目录号rms-sp)根据生产商指南使用具有10%胎牛血清的高葡萄糖dmem培养基进行培养。给所述培养基补充50u/ml青霉素、50ug/ml链霉素和100ug/ml normocin,以防止细菌污染。使用countess细胞计数器计算细胞密度和生存力,并除去适当体积的细胞以使每孔的接种密度为0.2x106个细胞/ml。将135ul细胞悬浮液与15ul对应的有效负载处理一起加入96孔平板中的每个孔中。一式三份运行每个测定点,并将平板在37℃和5% co2下温育24或72小时。为了评估nfκb诱导,通过将200ul的qb试剂(invivogen目录号rep-qbs)和200ul的qb缓冲液加入19.6ml水中来制备quanti-bluetm溶液。将所得溶液涡旋并在室温温育10分钟。将96孔平板在1990rpm离心10分钟,并将40ul细胞上清液加入160ul制备的quanti-bluetm溶液中。将qb反应板在37℃温育24小时。使用molecular devices i3x平板读数器在630nm的波长读出平板以确定seap产生量。在图1a和图1b中所示的数据表明,本发明的选定有效负载能够活化表达tlr7和tlr8的淋巴细胞细胞系中的nfκb信号传递途径。实施例14.评价针对表达mtlr7的hek293-报道细胞系的有效负载。表达小鼠tlr7的hek-bluetm mtlr7细胞系购自invivogen(目录号hkb-mtlr7)。将hek-bluetm mtlr7细胞维持在使用具有10%胎牛血清的高葡萄糖dmem培养基的培养基中。给所述培养基补充50u/ml青霉素、50ug/ml链霉素和100ug/ml normocin以防止细菌污染。在实验前,将hek-bluetm mtlr7细胞使用预热的dpbs冲洗并分离。将细胞收集并在1100rpm离心5分钟以除去上清液。然后将细胞以20万个细胞/ml接种悬浮液重新悬浮,并以90ul的接种体积接种到96孔平板中。将瑞喹莫德、e66和e104稀释至30、3、0.3和0.03um。将10ul有效负载溶液加入相应的孔中以达到3、0.3、0.03和0.003um的终浓度梯度。将10ul dpbs加入用于空白。一式三份地进行实验。将平板在37摄氏度/5% co2环境下温育24小时。收集上清液,并通过运行quanti-bluetm测定来检测nfκb活化。简而言之,将从invivogen购买的quanti-bluetm试剂按照生产商的方案重新配制为检测溶液。将180ul的quanti-bluetm检测溶液与20ul的上清液混合,并在37摄氏度/5% co2环境中温育4小时。然后使用spectramaxi3x微量培养板读数器检测在630nm的吸光度。使用graphpad prism 7软件对数据进行分析和绘图。在图2中所示的数据证明,本文公开的有效负载激动小鼠-tlr7途径。实施例15.在ramos blue报告物测定中,连接基的连接消除了tlr激动剂活性。在0.1m pbs ph 7.4中制备50mm半胱氨酸储备液。将10当量的50mm半胱氨酸加入1当量的连接基-有效负载中以产生2.50mm溶液。将反应物涡旋并在室温静置1小时以允许迈克尔加成发生。1小时后,取出一个等分试样并通过uplc-ms运行以确保完全迈克尔反应。使用0.1m pbs ph 7.4将反应物稀释至1mm。将1mm溶液在96孔平板中稀释至500um和50um,以提供在细胞悬浮液中50um和5um的终浓度。使用countess细胞计数器计算细胞密度和生存力,并除去适当体积的细胞以使每孔的接种密度为0.2x106个细胞/ml。将135ul细胞悬浮液与15ul对应的连接基-有效负载处理一起加入96孔平板中的每个孔中。一式三份运行每种浓度,并在37℃和5% co2下温育72小时。通过将200ul的qb试剂和200ul的qb缓冲液加入19.6ml水中,制备invivogen的quanti-bluetm溶液。将得到的溶液涡旋并在室温温育10分钟。将96孔平板在1990rpm离心10分钟。然后,将40ul细胞上清液加入160ul制备的quanti-bluetm溶液中并在37℃温育24小时。使用molecular devices i3x平板读数器在630nm的波长读出平板以确定seap产生量。在表2中所示的数据解释了连接基-有效负载的诱导nfκb活化的倾向。表2.该表表明本发明的连接基-有效负载对ramos-blue细胞中的nfκb途径的活化。实施例16.tlr激动剂adc对ramos-blue细胞中nfκb活性的活化。使用0.5x106个细胞/ml和72小时温育时间至1x106个细胞/ml和96小时温育的接种密度,优化使用adc的ramos blue测定。将adc在pbs中稀释至1000μg/ml和300μg/ml的浓度。使用countess细胞计数器计算细胞密度和生存力,并除去适当体积的细胞以使每孔的接种密度为1x106个细胞/ml。将80ul细胞悬浮液与20ul对应的adc处理一起加入96孔平板中的每个孔中。评价100ug/ml和30ug/ml的最终adc浓度。一式四份地执行每种测定条件并在37℃和5%co2下温育96小时。在选定的实施例中,将细胞用100ug/ml裸抗体的预剂量处理,并在adc处理之前在37℃和5% co2下温育15分钟。将细胞用10ul的裸抗体处理,然后在15分钟温育后用10ul的adc处理。一式四份地运行样品并温育96小时。通过将200ul的qb试剂和200ul的qb缓冲液加入19.6ml水中,制备invivogen的quanti-bluetm溶液。将得到的溶液涡旋并在室温温育10分钟。将96孔平板在1990rpm离心10分钟。然后,将40ul细胞上清液加入160ul制备的quanti-bluetm溶液中并在37℃温育24小时。使用molecular devices i3x平板读数器在630nm的波长读出平板以确定seap产生量。在表3中所示的数据表明,本发明的adc在b-细胞报告物测定中活化nfκb途径,从而表明作为选择性免疫调节剂的潜力。表3实施例17.来自靶向乳癌的adc的条件培养基能够活化mtlr7报告物系统。将过表达her2的乳癌细胞系skbr3维持在具有10%胎牛血清的高葡萄糖dmem培养基中。在实验前,将skbr3细胞胰蛋白酶化,离心,并以20万个细胞/ml接种悬浮液重新悬浮。然后将skbr3细胞(100ul,20,000个细胞)接种至96孔平板并允许粘附过夜。还如前面描述的,以20万个/ml的密度和90ul的体积将hek-bluetm mtlr7细胞接种至96孔平板。应用青霉素/链霉素和normocintm以防止微生物污染。将tlr7激动剂adc稀释至期望的浓度。将10ul的adc溶液、含有裸单克隆抗体的adc溶液或dpbs加入对应孔中并彻底混合。一式三份地进行实验。在处理以后,将skbr3细胞在37℃/5% co2环境中温育48小时。温育后,将hek-bluetmmtlr7平板的上清液用skbr3平板的对应孔的上清液替换。将hek-bluetm mtlr7细胞温育48小时,然后收集上清液,并通过在实施例14中描述的quanti-bluetm测定来测量nfκb水平。使用graphpad prism 7软件对数据进行分析和绘图。图3表明,在本发明中感兴趣的adc可以被表达抗原的肿瘤组织代谢,从而导致附近的表达tlr7的细胞的活化。这种活性可以通过联合给药裸抗体或通过靶向不表达的抗原来抑制。图4和图5表明,与瑞喹莫德adc相比,e104和e66 adc具有优异的活性。图6表明,替代连接基(消除pabc间隔基)在活化附近的tlr7细胞方面不那么有效。实施例18:来自巨噬细胞的细胞因子释放人单核细胞系thp-1购自atcc。如下完成thp-1向巨噬细胞的分化:将thp-1细胞接种至96孔平板(20,000个细胞/孔),并在100ul的rpmi 1640培养基中培养72小时,该培养基具有含有200nm佛波醇-12-肉豆蔻酸酯-13-乙酸酯(pma)的10%胎牛血清。温育后,除去上清液,并将细胞用预温热的dpbs冲洗两次以消除残留的pma。然后将细胞在具有10%胎牛血清的新鲜rpmi 1640培养基中培养至少72小时。在显微镜下证实巨噬细胞分化的形态学证据。为了试验来自thp-1和thp-1分化的巨噬细胞的细胞因子释放,将thp-1单核细胞以20万个/ml的密度和90ul的体积接种至96孔平板。从巨噬细胞平板除去旧培养基并添加90ul新鲜培养基。以150、30、6和1.2um制备有效负载的储备溶液。将10ul有效负载溶液加入相应的孔中并充分混合以达到15、3、0.6和0.12um的终浓度梯度。一式三份地进行实验,并使用青霉素/链霉素避免细菌污染。将10ul dpbs加入空白中以保持体积一致。将细胞在37摄氏度/5% co2下温育24小时。温育后,将平板离心,并收集上清液样品。使用duoset tnfαelisa试剂盒(r&d systems)检测在每个上清液样品中的tnfα水平。使用graphpad prism 7软件对数据进行分析和绘图。图21表明,本发明的化合物诱导巨噬细胞和单核细胞释放tnfα。从巨噬细胞释放的细胞因子的量明显高于单核细胞。实施例19:血清稳定性的评价。将100ug/ml的adc加入250ul的小鼠或人血清中并使用0.1m pbs ph 7.4调至400ul的终体积。通过加入150ul 0.1m pbs ph 7.4至250ul的小鼠或人血清来制备空白。将反应物在37℃和5% co2下温育7天。在时间0、6小时和1、2、4和7天取50ul等分试样,并立即在-80℃冷冻以防止进一步反应。将样品解冻并将20ul加入40ul acn中以诱导蛋白沉淀。将样品涡旋并以100,000rmp离心10分钟。取出40ul上清液并将其加入到40ul水中。通过在1:3acn:水中进行10倍系列稀释来制备标准曲线,以具有10um至0.1nm的范围。使用优化的mrm方法将10ul等分试样注入waters xevo tqd质谱仪中进行定量。瑞喹莫德的扫描的转变(transition)为314.40→152.02,197.05,251.09;e104的转变为346.19→105.95,197.96,241.00;e66的转变为360.25→92.25,119.93,和241.01。图8和图9显示了稳定性研究的结果。实施例20.从1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(e104)衍生出的另外的活化tlr7/8的有效负载的制备.将e104与boc-甘氨酸、boc-β-ala-oh、boc-g-氨基丁酸、boc-5-氨基戊酸和boc-6-氨基己酸偶联:20a.将1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(15.2mg,1当量,44.0μmol)溶解在dma(2.0ml)中并用diea(5.6mg,7.6μl,0.99当量,44μmol)和boc-甘氨酸(7.7mg,1.0当量,44μmol)处理。将edc(16.9mg,2当量,88.0μmol)加入溶液中,并将反应物在室温搅拌12小时。将反应物通过制备型hplc纯化以得到(2-((4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基)-2-氧代乙基)氨基甲酸叔丁酯。lcms rt=2.74min;m/z=503.6[m+h]。该一般程序也用于产生下述衍生物:20b.(3-((4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基)-3-氧代丙基)氨基甲酸酯。lcms rt=2.78min;m/z=517.7[m+h]20c.(4-((4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基)-4-氧代丁基)氨基甲酸叔丁酯。lcms rt=2.80min;m/z=531.6[m+h]20d.(5-((4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基)-5-氧代戊基)氨基甲酸叔丁酯。lcms rt=2.87min;m/z=545.6[m+h]20e.(6-((4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基)-6-氧代己基)氨基甲酸叔丁酯。lcms rt=2.90min;m/z=559.7[m+h]。20f.将boc-保护的物质(20a)溶解在dcm(400μl)中并用tfa(59g,40μl,56当量,0.52mmol)处理。搅拌1h以后,将反应物浓缩至干燥以得到期望产物2-氨基-n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)乙酰胺。lcms rt=0.72min;m/z=403.5[m+h]。该一般程序也用于产生下述衍生物:20g.3-氨基-n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)丙酰胺。lcms rt=2.78min;m/z=417.5[m+h]20h.4-氨基-n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)丁酰胺。lcms rt=2.81min;m/z=431.3[m+h]20i.5-氨基-n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)戊酰胺。lcms rt=3.42min;m/z=445.5[m+h]20j.6-氨基-n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)己酰胺。lcms rt=2.33min;m/z=459.5[m+h]。20k.将1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(15.0mg,1当量,43.4μmol)溶解在乙酸乙酯(400μl)中并用三乙胺(5.7mg,7.9μl,1.3当量,57μmol)处理。将反应物在冰浴上冷却。然后将戊酰氯(5.8mg,5.7μl,1.1当量,48μmol)加入预冷却的乙酸乙酯(133μl)中,随后将其逐滴转移至反应物。使反应物达到室温并搅拌过夜以后,通过与上面相同的方法将另外的1.3当量的三乙胺和另外的1.1当量的戊酰氯加入反应物中。将反应物通过制备型hplc纯化以得到期望的产物n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)戊酰胺。lcms rt=2.82min;m/z=430.6[m+h]。20l.将1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(10.5mg,1当量,30.4μmol)溶解在dcm(1.4ml)中。然后将乙酸酐(9.31mg,10.2μl,3当量,91.2μmol)加入反应混合物中并在室温搅拌3小时并通过hplc监测。然后将其通过制备型hplc纯化以得到2.8mg的期望的产物,n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)乙酰胺。lcms rt=2.71min;m/z=388.4[m+h]。20m.将1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(14.1mg,1当量,40.8μmol)溶解在dcm(2.0ml)中。将三乙胺(8.26mg,11.4μl,2当量,82μmol)和氯甲酸乙酯(8.9mg,7.9μl,2.0当量,82.0μmol)依次加入反应混合物中。将反应物在室温保持12小时,在该时间以后加入另外2当量的氯甲酸乙酯。搅拌另外1h以后,将反应物通过制备型hplc纯化以得到期望的产物,(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸乙酯。lcms rt=2.77min;m/z=418.5[m+h]。该一般程序也用于产生下述衍生物:20n.(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸异丙酯。lcms rt=2.91min;m/z=431.5[m+h]20o.2-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)乙酸丁酯。lcms rt=3.00min;m/z=446.6[m+h]。实施例21.3-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯酚的制备方案1:试剂(i)rnh2,net3,ch2cl2;(ii)zn,hcoonh4,meoh;(iii)c4h9cocl,net3,etoac;(iv).naoh,etoh:水(13:2);(v)a.mcpba,chcl3;b.nh4oh/nh3,chcl3;(vi).bbr3,ch2cl2步骤1.21a.n-(3-甲氧基苄基)-3-硝基喹啉-4-胺:向4-氯-3-硝基喹啉(1.500g,1.0当量,7.20mmol)在dcm(22.0ml)中的悬浮液中加入(3-甲氧基苯基)甲胺(986mg,0.92ml,1.0当量,7.20mmol)和三乙胺(1.09g,1.50ml,1.50当量,10.8mmol)。将混合物在40℃回流1h。通过uplc监测反应进程。在60min内实现反应物向期望的产物的完全转化,形成亮黄色悬浮液。将反应混合物冷却至室温,用水洗涤,过滤并将固体在真空下干燥以得到作为亮黄色粉末的期望的化合物(2.18g)。lc/ms rt=3.25min;m/z=310.1[m+h]。步骤2.21b.n4-(3-甲氧基苄基)喹啉-3,4-二胺:向步骤1的产物n-(3-甲氧基苄基)-3-硝基喹啉-4-胺(2.179g,1.0当量,7.04mmol)在meoh(25.0ml)中的悬浮液中加入锌(1.490g,4.0当量,28.0mmol)和氯化铵(1.80g,4.0当量,28.0mmol)。将反应混合物在室温搅拌10min(以产生灰色悬浮液)并通过uplc监测。产物立即开始形成。10分钟以后,将反应混合物穿过硅藻土过滤并将溶剂在真空中蒸发。将残余物溶解在dcm中,在1m naoh中分配,用10%meoh在dcm中的溶液(50mlx3)洗涤并经mgso4干燥。在真空下除去溶剂以得到1.36g(70%)作为黑棕色油的标题化合物。lc/ms rt=2.44;m/z=280.1[m+h]。步骤3.21c.n-(4-((3-甲氧基苄基)氨基)喹啉-3-基)戊酰胺:向冷却至0℃的在无水etoac(45.0ml)中的步骤2的粗产物n4-(3-甲氧基苄基)喹啉-3,4-二胺(1.363g,1.0当量,4.88mmol)中加入预先冷却的三乙胺(642mg,884μl,1.3当量,6.34mmol)。将反应物在室温搅拌5分钟。此后,在0℃逐滴加入在etoac(15.0ml)中的戊酰氯(883mg,868μl,1.5当量,7.32mmol),并将反应混合物进一步搅拌15min,并通过uplc监测。将反应物用乙醇淬灭,然后在减压下浓缩,形成黄褐色晶体。lc/ms rt=2.56min;m/z=364.4[m+h]。步骤4.21d.2-丁基-1-(3-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉:将步骤3的粗产物n-(4-((3-甲氧基苄基)氨基)喹啉-3-基)戊酰胺(1.770g,1.0当量,4.88mmol)溶解在etoh(26.0ml)中并用在h2o(4.00ml)中的氢氧化钠(464mg,1.0当量,11.6mmol)处理。将反应混合物在80℃回流24h并通过uplc监测进程。结束后,将溶液溶解在水(75ml)中并在etoac(75ml)中分配。通过用etoac(75mlx3)洗涤水层来萃取所述物质。将有机层经mgso4干燥并蒸发至干燥。将干燥的琥珀色粗萃取物(1700mg)使用10% meoh在dcm中的溶液通过硅胶色谱法纯化。回收的纯物质为琥珀色树脂(856mg,51%)。lc/ms rt=2.97min;m/z=346.2[m+h]。1hnmr(400mhz,dmso)δh/ppm 9.21(s,1h),8.11(d,j=8.5hz,2h),7.61(t,j=7.7hz,1h),7.50(t,j=7.7hz,1h),7.21(t,j=8.0hz,1h),6.83(d,j=8.2hz,1h),6.66(s,1h),6.49(d,j=7.7hz,1h),5.93(s,2h),3.67(s,3h),2.96(t,j=7.5hz,2h),1.78(五重峰,j=7.5hz,2h),1.40(六重峰,j=7.4hz,2h),0.88(t,j=7.4hz,3h)。21e.步骤5a)2-丁基-1-(3-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉-5-氧化物:将在chcl3(2.5ml)中的2-丁基-1-(3-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉(156mg,1.0当量,450mmol)用3-氯过氧苯甲酸(173mg,1.6当量,700mmol)处理并在40℃搅拌1h。将剩余物质不经进一步纯化或分离用于下一步。lc/ms rt=3.26m/z=362.4[m+h]。步骤5b)2-丁基-1-(3-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉-4-胺:在50℃向含有在chcl3(2.5ml)中的2-丁基-1-(3-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉-5-氧化物(162.8mg,1当量,450umol)的前一个反应物中逐滴加入28-38%氢氧化铵2.2g,2.5m l,28当量,13mmol),随后加入4-甲基苯磺酰氯(174mg,2当量,920umol)。将混合物在室温搅拌1h并通过uplc监测。结束后,将反应物用氯仿和碳酸氢盐水溶液萃取,经mgso4干燥并在真空中除去溶剂以得到183mg(112%)的粗产物。将物质不经进一步纯化继续用于形成3-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯酚。通过制备型hplc纯化级分,提供作为白色残余物的标题化合物。lc/ms rt=2.78min;m/z=361.2[m+h]。1h nmr(400mhz,dmso)δh/ppm 7.97(d,j=7.9hz,1h),7.80(d,j=7.8hz,1h),7.63(t,j=7.8hz,1h),7.38(t,j=7.8hz,1h),7.24(t,j=8.0hz,1h),6.86(d,j=8.2,1h),6.71(s,1h),6.54(d,j=8.0hz,1h),5.93(s,2h),3.70(s,3h),2.97(t,j=7.7hz,2h),1.72(五重峰,j=7.6hz,2h),1.38(六重峰,j=7.4hz,2h),0.87(t,j=7.4hz,3h)。步骤6.21f.3-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯酚:在氮气下向冷却至0℃的粗萃取物2-丁基-1-(3-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉-4-胺(105.5mg,1.0当量,293μmol)在dcm(1.00ml)中的悬浮液中逐滴加入在dcm(0.70ml)中的bbr3(220.0mg,83ul,3.0当量,878μmol)。将混合物在0℃搅拌5分钟,然后达到室温并通过uplc监测1.5h。结束后,将反应物通过倒在冰水上进行淬灭。将该溶液用dcm和碳酸氢盐水溶液萃取,将碳酸氢盐相用dcm(10mlx2)洗涤,经mgso4干燥,并蒸发至干燥。将残余物通过制备型hplc纯化,提供作为白色细粉末的标题化合物(35.0mg,35%)lc/ms:保留时间=2.57min:m/z347.2[m+1]。1h nmr(400mhz,dmso)δh/ppm 7.97(d,j=7.8hz,1h),7.80(d,j=8.0hz,1h),7.64(t,j=7.8hz,1h),7.39(t,j=7.8hz,1h),7.14(t,j=7.8hz,1h),6.66(d,j=8.1hz,1h),6.52(d,j=7.9hz,1h),6.39(s,1h),5.88(s,2h),2.97(t,j=7.7hz,2h),1.72(五重峰,j=7.6hz,2h),1.39(六重峰,j=7.4hz,2h),0.87(t,j=7.3,3h)实施例22:4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯酚的制备步骤1:22a.将4-氯-3-硝基喹啉(1003.4mg,1当量,4.8102mol)溶解在dcm(15.0ml)中并用(4-甲氧基苯基)甲胺(725mg,690μl,1.10当量,5.28mmol)处理,并然后用三乙胺(973.48mg,1.34ml,2当量,9.6203mmol)处理。将反应物加热至30℃并搅拌1小时,同时通过uplc监测。然后将反应物冷却,浓缩至干燥,并与水一起研磨。将产物通过真空过滤分离,产生作为亮黄色粉末的n-(4-甲氧基苄基)-3-硝基喹啉-4-胺(1249.4mg,4.0391mmol,83.969%收率)。lcms rt=3.23min;m/z=310.1[m+h]。步骤2:22b.将n-(4-甲氧基苄基)-3-硝基喹啉-4-胺(501.2mg,1当量,1.620mmol)悬浮于甲醇(30.0ml)中。将溶液搅拌并加入氯化铵(871.8mg,10.06当量,16.30mmol)。然后将其放入冰浴中并用锌(1062.8mg,10.03当量,16.30mmol)处理。将反应物保持共计25分钟并通过uplc监测,产生534.3mg的n4-(4-甲氧基苄基)喹啉-3,4-二胺粗产物。将粗产物溶解在1:1甲醇:dcm中并穿过硅藻土过滤以便除去过量的锌。然后将溶剂蒸发并将产物再溶解在10%的甲醇在dcm中的溶液(40.0ml)中,并用1m naoh(40.0ml)洗涤。将此重复3次,并然后将有机层使用mgso4干燥。然后将其干燥以回收n4-(4-甲氧基苄基)喹啉-3,4-二胺(307.6mg,57.6%回收率)。lcms rt=1.64min;m/z=280.4[m+h]。步骤3和4:22d.继续使用307.6mg的n4-(4-甲氧基苄基)喹啉-3,4-二胺并溶解在乙酸乙酯(5ml)中。加入三乙胺(144.9mg,200μl,1.3当量,1.432mmol)并将反应物在冰上冷却。将戊酰氯(152mg,150μl,1.15当量,1.26mmol)与1.25ml冷却的乙酸乙酯组合,然后将其逐滴加入反应混合物中。将得到的混合物在冰浴中搅拌20分钟,同时通过hplc监测反应。结束后,将反应物浓缩至干燥,达到575.7mg的最终重量。lcms rt=2.59min;m/z=364.4[m+h]。然后将n-(4-((4-甲氧基苄基)氨基)喹啉-3-基)戊酰胺的粗产物(575.5mg)溶解在乙醇(13ml)中。在一个单独的容器中,将naoh(127.4mg,2.893当量,3.185mmol)溶解在水(1.8ml)中。将反应物和naoh溶液加热至80℃。然后将氢氧化钠加入反应物中。将反应物在80℃回流加热3.5小时并通过uplc监测。然后将25ml水加入反应物中并将其在dcm中分配。将有机层用水洗涤3次,并然后用硫酸镁干燥。将粗制物质(342.4mg)使用0%->10%的甲醇在dcm中的梯度使用硅胶色谱法进一步纯化。将分离的级分浓缩至干燥,产生153.8mg回收的纯的2-丁基-1-(4-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉(44.9%)。lcms rt=2.95min;m/z=346.3[m+h]。步骤5:22e.将2-丁基-1-(4-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉(141.4mg,1当量,409.3μmol)溶解在氯仿(2.5ml)中并用3-氯过氧苯甲酸(215.6mg,3.052当量,1.249mmol)处理。将混合物涡旋并加热至40℃并搅拌1小时,导致2-丁基-1-(4-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉5-氧化物的形成。lcms rt=3.24min;m/z=362.3[m+h]。将反应物冷却回室温并然后剧烈搅拌,然后加入氢氧化铵(405mg,460μl,28.2当量,11.5mmol)。搅拌1h以后,将4-甲基苯磺酰氯(161.2mg,2.006当量,845.6μmol)加入反应物中。30分钟以后,将反应物浓缩至干燥并再溶解在40ml氯仿中。将有机溶液用40ml碳酸氢钠洗涤,随后用40ml盐水洗涤,产生229.2mg回收的粗产物。然后将该粗制物质的一部分(70.6mg)通过制备型hplc进一步纯化,产生31.9mg的纯的甲氧基e104物质,2-丁基-1-(4-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉-4-胺。lcms rt=2.77min;m/z=361.5[m+h]。步骤6:22f.将23.5mg的2-丁基-1-(4-甲氧基苄基)-1h-咪唑并[4,5-c]喹啉-4-胺溶解在dcm(500μl)中并放在氮气下。在冰浴中使温度降低。将20μl的bbr3用500μl的dcm稀释并然后将其逐滴加入反应物中。将反应物温热至室温并放置2小时,同时通过hplc监测。将反应物在室温放置过夜,该时间以后加入另外3当量的bbr3并将反应物搅拌另外2h。将混合物小心地用1ml水和1ml碳酸氢钠淬灭。将有机层经硫酸镁干燥,产生5.2mg不纯的物质,然后将其通过制备型hplc进一步纯化,产生3.3mg的纯的4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯酚。lcms rt=2.13min;m/z=347.4[m+h]。实施例23:1-(3-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺的制备方案1:试剂(i)rnh2,net3,ch2cl2;(ii)zn,hcoonh4,meoh;(iii)c4h9cocl,net3,etoac;(iv)naoh,etoh:水(13:2);(v)a.mcpba,chcl3;b.nh4oh/nh3,chcl3;(vi)20%tfa,ch2cl2步骤1.23a.(3-(((3-硝基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯:向(3-(氨基甲基)苯基)氨基甲酸叔丁酯(1077mg,1当量,4.85mmol)在dcm(27.0ml)中的溶液中加入4-氯-3-硝基喹啉(1005mg,1当量,4.82mmol)和三乙胺(975mg,1.34ml,2当量,9.64mmol)。混合物为棕色,并且一旦加入三乙胺就变为黄色。将混合物在40℃回流1h。通过hplc监测反应进程。在60min内实现反应物向期望的产物的接近完全转化,形成黄色沉淀物。将反应物冷却至室温,并在减压下浓缩至干燥,形成黄色晶体。将晶体悬浮于水(140ml)中,在真空下穿过烧结漏斗过滤并在干燥器中干燥以得到期望的化合物(1550.2mg,81.54%收率)hplc rt=3.50min(极性梯度);m/z=394.43[m+h]。步骤2.23b.(3-(((3-氨基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯:将(3-(((3-硝基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯(650mg,1当量,1.65mmol)在meoh(20ml)中的悬浮液用预冷却的锌(1099mg,10.2当量,16.81mmol)和氯化铵(899.8mg,10.2当量,16.82mmol)在meoh(6ml)中的悬浮液处理。将混合物在0℃搅拌20min并通过hplc监测。混合物快速地转变为灰色/绿色悬浮液。20min以后,将反应混合物穿过硅藻土过滤并将溶剂在真空中蒸发。将残余物溶解在dcm中,用水洗涤,并反萃取进dcm中。将有机层经mgso4干燥。在真空下除去溶剂以得到期望的化合物(549.2mg,91.4%)。hplc rt=2.66min(极性梯度);m/z=365.45[m+h]。步骤3.23c.(3-(((3-戊烷酰氨基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯:将冷却至0℃的在无水etoac(16ml)中的粗制的(3-(((3-氨基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯(549.2mg,1.0当量,1.51mmol)加入也在0℃的三乙胺(198.2mg,273μl,1.3当量,1.96mmol)中。将该混合物搅拌10min。接着,在0℃将在etoac(5ml)中的戊酰氯(236.2mg,232.5μl,1.3当量,1.96mmol)逐滴加入反应混合物中。将混合物搅拌40min并通过hplc监测。将溶液在减压下浓缩至干燥并不经纯化地继续使用。hplc rt=2.79min(极性梯度)m/z=449.57[m+h]。步骤4.23d.(3-((2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸叔丁酯:将来自前一步的粗产物((3-(((3-戊烷酰氨基喹啉-4-基)氨基)甲基)苯基)氨基甲酸叔丁酯,676mg,1当量,1.51mmol)溶解在etoh(9ml)中并用在h2o(1.38ml)中的氢氧化钠(121mg,2当量,3.01mmol)处理。将混合物回流4h并通过hplc监测。将水直接加入反应溶液中并将产物用dcm萃取。将有机层经mgso4干燥并蒸发至干燥。将琥珀色-红色产物使用硅胶色谱法纯化。使用100%至5% meoh在dcm中的溶液的11min梯度,将得到的洗脱液在减压下浓缩至干燥(647.3mg,99.8%)hplc rt=3.15min(极性梯度)m/z=431.55[m+h]。步骤5.23e.a.1-(3-((叔丁氧基羰基)氨基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉5-氧化物:将3-氯过氧苯甲酸(382mg,2.896当量,1.55mmol)加入(3-((2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸叔丁酯(230.4mg,1.0当量,0.535mmol)在chcl2(3ml)中的溶液中。将反应混合物在40℃搅拌1h。将粗制的溶液在空气流下干燥并不经后处理继续用于下一步。hplc rt=3.50min(极性梯度)m/z=447.55[m+h]。b.(3-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸叔丁酯:将来自前一步的1-(3-((叔丁氧基羰基)氨基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉5-氧化物(239.0mg,1当量,.535mmol)再溶解在chcl3(10ml)中并温热至50℃。逐滴加入氢氧化铵(20%)(2.81g,30当量,16.0mmol)并将混合物在50℃搅拌1h。将4-甲基苯磺酰氯(204.1mg,2当量,1.070mmol)加入该混合物中并将得到的反应物在50℃搅拌4h。然后将反应物用chcl3(40ml)稀释并将有机级分用碳酸氢盐水溶液洗涤,然后用盐水洗涤。将有机层用mgso4干燥在减压下干燥。然后将粗产物通过硅胶色谱法(0→20% dcm/meoh)纯化。将纯的级分在减压下干燥,产生164.7mg(69.1%)标题化合物。hplc rt=2.94min(极性梯度)m/z=446.57[m+h]。步骤6.23f.1-(3-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺:向(3-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基甲酸叔丁酯(30mg,67μmol)在dcm中的溶液中加入tfa(78μl,15当量)。将反应混合物在23℃搅拌3h。将得到的产物混合物在减压下干燥,产生作为深红色-琥珀色晶体的期望物质(16.3mg,71%)hplc rt=2.61min(极性梯度)m/z=346.45[m+h]。实施例24:活化trl的连接基-有效负载的制备。(3-((4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基)-3-氧代丙基)氨基甲酸4-((s)-2-((s)-2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯(24a):将3-氨基-n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)丙酰胺(14.2mg,1当量,34.1μmol)溶解在dma(1500μl)中。然后加入mcvalcitpabc-pnp(25.2mg,1当量,34.1μmol)、2,6-二甲基吡啶(7.31mg,2当量,68.2μmol)和hobt(6.3mg,1.2当量,41μmol)。将其在室温搅拌几天并通过hplc监测。然后将其通过制备型hplc纯化以得到10.3mg标题产物。hplc rt=2.79min,m/z=1015.8[m+h]。n-(3-((4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基)-3-氧代丙基)-6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰胺(24b):将3-氨基-n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)丙酰胺(14.7mg,1当量,35.3μmol)溶解在dma(1500μl)中并加入hobt(7.7mg,1.4当量,50μmol)、2,6-二甲基吡啶(11.3mg,12.3μl,3当量,106μmol)和6-马来酰亚胺基己酸(7.5mg,1当量,36μmol)。然后最后加入hatu(17.4mg,1.3当量,45.9μmol)。将其在室温搅拌1.5小时并通过hplc监测。然后将其通过hplc纯化以得到7.6mg的标题产物。hplc rt=2.96min,m/z=610.5[m+h]。上述一般程序也用于产生:(1-(4-(3-((叔丁氧基羰基)氨基)丙烷酰氨基)苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-基)氨基甲酸4-((s)-2-((s)-2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯(24c)lcms rt=3.10min;m/z=1116.3[m+h](2-丁基-1-(4-戊烷酰氨基苄基)-1h-咪唑并[4,5-c]喹啉-4-基)氨基甲酸4-((s)-2-((s)-2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)-3-甲基丁酰氨基)-5-脲基戊烷酰氨基)苄酯(24d)lcms rt=3.15min;m/z=1029.2[m+h]。通过上述程序的轻微修改来制备另外的类似物诸如24e-24i(下面)。实施例25:(s)-n1-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)-2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)琥珀酰亚胺(mcasn-e104)的制备步骤1:(s)-(1-((4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)氨基)-1,4-二氧代-4-(三苯甲基氨基)丁烷-2-基)氨基甲酸叔丁酯:向4ml玻璃lc/ms管形瓶中,加入750ul的dmf,随后加入20mg的1-(4-氨基苄基)-2-丁基-1h-咪唑并[4,5-c]喹啉-4-胺(e104)(1.4当量)、20mg的boc-asn(trt)-oh(1当量)、10mg的hobt(1当量)和28mg的hatu(1.8当量)。短暂涡旋以后,将dipea(4当量)加入以开始反应。溶液为澄清黄色并允许静置30小时,并然后使用水和acn+tfa流动相和标准梯度通过制备型hplc纯化,产生4.3mg(9.3%)标题化合物。步骤2:将来自步骤1的物质用950ul的tfa处理,反应物立即变成鲜艳黄色。2分钟以后,加入三乙基硅烷,导致黄色的消散。3分钟以后,lc/ms指示去保护是完全的。将产物风干以除去tfa并直接用在下一步中。步骤3:(s)-n1-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)-2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)琥珀酰亚胺:将来自步骤2的粗制物质溶解在500ul的dmf中并转移至1ml玻璃lc/ms管形瓶。加入1.5当量的mcosu,随后加入10当量的dipea(以中和过量的tfa)。运行lc/ms以监测反应进程。将标题产物使用acn+水(含tfa添加剂)在标准梯度下通过制备型hplc纯化,产生3.2mg(90%)的标题化合物。通过将所述固体溶解在约950ul的dma中来制备储备溶液,形成5mm储备溶液。hplc rt=2.41min;m+h=774.5da。通过上述程序的轻微修改来制备另外的类似物诸如25d、25e、25f、25g和25h(下面)。实施例26:另外的tlr激动剂连接基-有效负载的制备(2-丁基-1-(4-己烷酰氨基苄基)-1h-咪唑并[4,5-c]喹啉-4-基)氨基甲酸4-((s)-2-((s)-2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己烷酰氨基)丙烷酰氨基)丙烷酰氨基)苄酯:将激动剂(n-(4-((4-氨基-2-丁基-1h-咪唑并[4,5-c]喹啉-1-基)甲基)苯基)己酰胺)溶解在dma中并随后用2,6-二甲基吡啶(2当量)、hobt(1当量)和mcvalcitpab-pnp处理。在室温搅拌过夜以后,通过制备型hplc分离期望的产物。通过该方法的微小修改可以制备所示的另外产物(26a、26b、26c、26d、26e)。实施例27:连接基连接位点的证实。进行各种连接基-有效负载的lcms/ms片段化分析,以建立连接基添加的区域化学。下面说明了一个这样的例子。使用waters tqd lcms系统在“子离子扫描”模式下将获得的结构(按质量将其假定为异构体i或异构体ii)片段化。明显观察到片段iii(m/z=241),而未观察到片段iv(m/z=548)。据此,得出的结论是,母体结构由化合物i组成。nmr分析支持该结构确定。实施例28:使用位点特异性的硫醇化将有效负载连接至q295残基来制备新的adc:去糖基化:将1mg的igg1抗体(如下面所示)用4μl(2ug)的pngase f(bulldog bio)处理并用pbs稀释至500ul。将反应物在37℃温育过夜或直到通过lcms确定去糖基化结束。硫醇化:观察到完全去糖基化以后,加入240ul的0.1m sorensen氏磷酸盐缓冲液(ph 6),随后加入20ul的30mm胱胺水溶液(100当量)和70mg转谷氨酰胺酶粉末(ajinomoto)。将混合物彻底涡旋,轻轻加热直到溶液变成同质的。蛋白a捕获:在37℃温育48h以后,将物质加入已经在pbs中预平衡的蛋白a柱中。二硫化物的选择性还原:将树脂用3x600 ul的pbs洗涤,并然后用100ul含有10当量的tppms(又名二苯基膦基苯-3-磺酸钠)的pbs处理。在室温温育2h后,将树脂离心以除去过量的tppms。与适当的lp缀合:将树脂用100ul含有12当量的适当的连接基-有效负载的pbs处理。(应当指出,将连接基-有效负载作为在dma中的10mm储备溶液进行储存。)短暂涡旋以后,允许树脂在室温静置过夜。[应当指出,在某些情况下,添加另外的dmso以防止连接基-有效负载的沉淀。]洗脱:将树脂用600ul的pbs洗涤2次,并然后用400ul的甘氨酸缓冲液(ph 4)处理。静置3分钟以后,将树脂离心至含有30ul的tris缓冲液(ph 7.8)的试管中。再次重复洗脱步骤以确保adc的完全洗脱。将合并的洗脱液缓冲液交换进1ml的pbs中并过滤灭菌以供储存。表--通过以上方法制备的adc: 抗体 连接基-有效负载 最终的dar 质量变化 理论质量变化 %聚集 抗-her2 mce104(lp#11) 1.6 535 537 na 抗-her2 mcvalcitpabc-e104(lp#9) 2.0 946 944 na 抗-gcc mce104(lp#11) 1.6 539 537 na 抗-gcc mcvalcitpabc-e104(lp#9) 2.0 946 944 na 抗-trop2 mce104(lp#11) 1.7 543 537 na 抗-trop2 mcvalcitpabc-e104(lp#9) 2.0 943 944 na 抗-rsv mce104(lp#11) 1.7 541 537 na 抗-rsv mcvalcitpabc-e104(lp#9) 2.1 946 944 na 实施例29:在小鼠异种移植物模型中证实效力在sci/米色小鼠中进行乳癌异种移植物研究,以便确定adc的效用和靶向能力。将人乳癌细胞(hcc1954,atcc)在具有10% fbs(avantar)的roswell park memorialinstitute(rpmi)1640(corning)中培养,并维持在生产商推荐的密度下。应用100u ml-1青霉素-链霉素以防止微生物污染。在肿瘤植入前,将所述细胞胰蛋白酶化,冲洗,并重新悬浮在4000万个细胞/ml的悬浮液中。在即将植入之前,将悬浮液与matrigel(corning)以1:1混合以形成最终的植入混合物,将其在冰上保存不超过2小时。将大约200万个细胞(100μl混合物)皮下植入6-8周龄雌性scid/米色小鼠(charles river labs)的右胁腹。使用卡尺每周两次记录肿瘤体积并使用以下公式估计:长度×宽度2/2。一旦肿瘤体积达到50-300mm3,就开始治疗。将小鼠随机分配到10个不同的治疗组(每组5只小鼠)。通过腹膜内注射,共3次,间隔5天,给小鼠施用adc(10mg kg-1或3mg kg-1)、裸抗-her2 mab(10mg kg-1)或dpbs。每2-3天测量并记录肿瘤体积。根据iacuc批准的动物方案,对其肿瘤超过1000mm3、遭受溃疡形成或在研究期间表现出任何应激迹象的小鼠实施安乐死。结果显示在图10中。简言之,使用靶向的(抗-her2)adc的治疗导致快速肿瘤消退,而使用对应的非靶向的(抗-cd20)adc的治疗则不然。对于任何治疗组,未观察到体重的显著变化,从而提示adc被良好耐受(图11)。实施例30:在hek-bluemtlr7、htlr7、mtlr8和htlr8细胞中评价有效负载。将hek报道细胞系(invivogen)维持在使用具有10%fbs的高葡萄糖dmem培养基的培养基中,所述培养基补充了50u/ml青霉素、50ug/ml链霉素和100ug/ml normocin以防止细菌污染。在实验前,将细胞使用预热的dpbs冲洗并分离,并通过在1100rpm离心5min进行收集。然后将细胞以20万个细胞/ml重新悬浮并接种到96孔平板(90ul)中。将化合物在pbs中稀释至最终期望的浓度的10倍。将10μl有效负载溶液(或pbs空白)加入每个孔中。一式三份进行每个实验。将平板在37℃/5% co2环境下温育24h。收集上清液,并使用quanti-bluetm底物按照生产商的方案通过测量在630nm或650nm的吸光度来确定seap诱导(nfκb活化)。如图12-15所示的结果指示,本发明的选定化合物是针对人和小鼠同种型的有效的且选择性的tlr7激动剂,同时观察到弱至中等的tlr8活性。实施例31:对于另外的tlr7激动剂评价在ramos-blue细胞中的nfkb活化。将每种有效负载在10% dmso在pbs中的溶液中进行3倍系列稀释,以具有1000um至76nm的最终浓度范围。使用具有10%胎牛血清的高葡萄糖dmem培养基根据生产商指南培养ramos-blue细胞(invivogen,目录号rms-sp)。给所述培养基补充50u/ml青霉素、50ug/ml链霉素和100ug/ml normocin以防止细菌污染。使用countess细胞计数器计算细胞密度和生存力,并除去适当体积的细胞以使每孔的接种密度为0.2x106个细胞/ml。将135ul细胞悬浮液与15ul对应的有效负载处理一起加入96孔平板中的每个孔中。一式三份运行每个测定点,并将平板在37℃和5% co2下温育24或72小时。为了评估nfκb诱导,通过将200ul的qb试剂(invivogen目录号rep-qbs)和200ul的qb缓冲液加入19.6ml水中来制备quanti-bluetm溶液。将得到的溶液涡旋并在室温温育10分钟。将96孔平板以1990rpm离心10分钟,并将40ul细胞上清液加入160ul制备的quanti-bluetm溶液中。将qb反应板在37℃温育24小时。使用moleculardevices i3x平板读数器在630nm的波长读出平板以确定seap产生量。下表说明了导致seap背景(未处理)信号加倍的每种化合物的最低浓度。如所示的,本发明的化合物在从低nm至低微摩尔的范围内。na=无活性实施例32.使用抗-trop2和抗-gcc抗体将tlr激动剂递送至胰腺癌细胞系bxpc3和非小细胞肺癌细胞系a549。进行共培养实验,其中将a549(非小细胞肺癌)和小鼠巨噬细胞raw dual(invivogen)的各5000个细胞在37℃在5% co2气氛下在dmem高葡萄糖/10% fbs+青霉素/链霉素中培养。将细胞在各种浓度的选定adc存在下培养48h。取出培养基的20ul等分试样并将其加入到180ul的quanti-bluetm溶液(invivogen)中。温育4h或24h后,在630nm的吸光度指示在该巨噬细胞细胞系中nfκb途径的活化。结果显示在图16中。结果提供了本发明的抗-trop2和抗-gcc adc能够模拟在表达抗原的非小细胞肺癌组织附近的巨噬细胞的证据。使用bxpc3细胞(胰腺癌)进行了类似的实验,同样与小鼠巨噬细胞细胞系rawdual一起共培养。结果显示在图17中。结果提供了本发明的抗-trop2和抗-gcc adc能够模拟在表达抗原的胰腺癌组织附近的巨噬细胞的证据。实施例33.活化tlr的adc对表达her2的乳癌细胞无毒。在测定前,将skbr3和hcc1954细胞培养在具有10%胎牛血清的rpmi1640培养基中。简而言之,收获skbr3和hcc1954细胞并将其重新悬浮在20万个细胞/ml的接种悬浮液中。然后将90μl悬浮液接种到96孔平板中。将adc(抗-her2_mce104和抗-her_mcvalcitpabc_e104)稀释成浓度梯度(3倍系列稀释,10种不同浓度,包括0),并将10μl的adc溶液加入到相应的孔中并轻轻混合。然后将平板在37℃/5% co2下温育72小时。温育后,使用xtt细胞生存力测定试剂盒(biotium)获得细胞生存力。在graphpad prism软件中使用“log[抑制剂]相对于应答-可变斜率(四个参数)”方程生成ic50曲线。证实了两种adc对hcc1954和skbr3细胞的ic50均>30μg/ml。该数据表明,观察到的体内效力(实施例29)不是由于直接毒性,而是由于附近的肿瘤相关的淋巴细胞的间接活化。可以在下文中描述本发明的各种优选实施方案[a]至[aq]:[实施方案a]式(i)或(ii)的化合物其中:r1选自c1-c10烷基、c1-c10氧杂烷基和c1-c10氮杂烷基;r2和r3各自独立地选自氢、c1-c5烷基和c1-c5烷氧基;n是1或2y独立地选自任选地被取代的芳基和任选地被取代的杂芳基;z1选自-nrz-、-o-、-nrzc(o)-、-nrzc(o)-o-和-nrzso2-;z2不存在,或选自(c1-c8)烃-nh-和5-8元含氮杂环,其中所述杂环的氮连接至x2;z独立地选自-nrz-、-nrzc(o)-和-o-;rz在每种情况下独立地选自氢、c1-c8烃、c1-c8氧杂烷基、c1-c8氮杂烷基、杂芳基和5-8元杂环;x1独立地选自rz、-c(o)-rz、-c(o)-o-rz、-c(o)-n-(rz)2、-(ch2)knrzc(o)-(c1-c6)烷基、-(ch2)knrzc(o)-o-(c1-c4)烷基和-so2-rz;k是1至8的整数;x2包含可切割的或不可切割的连接基;且ab包含抗体或抗体片段。[实施方案b]式(i)或(ii)的上面实施方案[a]或根据本发明的其它实施方案的化合物,其中:r1选自c1-c10烷基、c1-c10氧杂烷基和c1-c10氮杂烷基;r2和r3各自独立地选自氢、c1-c5烷基和c1-c5烷氧基;n是1或2y独立地选自任选地被取代的芳基和任选地被取代的杂芳基;z1选自-nrz-、-o-、-nrzc(o)-、-nrzc(o)-o-和-nrzso2-;z2不存在,或选自(c1-c8)烃-nh-和5-8元含氮杂环,其中所述杂环的氮连接至x2;z选自-nrz-和-o-;rz在每种情况下独立地选自氢、c1-c8烃、c1-c8氧杂烷基、c1-c8氮杂烷基、杂芳基和5-8元杂环;x1独立地选自rz、-c(o)-rz、-c(o)-o-rz、-c(o)-n-(rz)2和-so2-rz;x2包含可切割的或不可切割的连接基;且ab包含抗体或抗体片段。[实施方案c]上面实施方案[a]或[b]中的任一个或根据本发明的其它实施方案的化合物,其中z2不存在,或选自-(c1-c8)烷基-nh-、-苄基-nh-、苯基-nh和5-8元含氮杂环。[实施方案d]上面实施方案[a]至[c]中的任一个或根据本发明的其它实施方案的化合物,其中:x2是l1-l2-(l3)p-(l4)q-(l5)r;l1是缀合部分;l2是间隔基单元,其选自分支的或未分支的c1-c12烷基、选自peg1至peg12的peg、l3是1至6个氨基酸的肽;l4是自降解的间隔基;l5是羰基;且p、q和r各自独立地选自0和1,其中当p和q各自是0时,r必须是0。[实施方案e]上面实施方案[a]至[d]中的任一个或根据本发明的其它实施方案的化合物,其中所述化合物具有式(i),且:r1选自正丁基、-ch2oh和-ch2och2ch3;r2和r3各自是氢;n是1;y是苯基或吡啶基,其中每个是未被取代的或被卤素、c1-c4烷基、c1-c4烷氧基、c1-c4卤代烷基或c1-c4卤代烷氧基中的一种或多种取代;x1是氢;且z1是-n(rz)-或-o-。[实施方案f]上面实施方案[a]至[d]中的任一个或根据本发明的其它实施方案的化合物,其中所述化合物具有式(ii),且:r1选自正丁基、-ch2oh和-ch2och2ch3;r2和r3各自是氢;n是1;y是苯基或吡啶基,其中每个是未被取代的或被卤素、c1-c4烷基、c1-c4烷氧基、c1-c4卤代烷基或c1-c4卤代烷氧基中的一种或多种取代;x1是氢;且z是-n(rz)-或-o-。[实施方案g]上面实施方案[a]或[b]中的任一个或根据本发明的其它实施方案的化合物,其中所述式(i)的化合物是式(ia)、(ib)、(ic)、(id)、(ie)、(if)、(ig)或(ih)的化合物:其中代表与x2的连接点。[实施方案h]上面实施方案[a]、[b]或[g]中的任一个或根据本发明的其它实施方案的化合物,其中所述式(i)的化合物是式(ia)、(ic)、(ie)或(ig)的化合物。[实施方案i]上面实施方案[a]、[b]、[g]或[h]中的任一个或根据本发明的其它实施方案的化合物,其中所述式(i)的化合物是式(ia)的化合物。[实施方案j]上面实施方案[a]或[b]中的任一个或根据本发明的其它实施方案的化合物,其中所述式(ii)的化合物是式(iia)、(iib)、(iic)、(iid)、(iie)、(iif)、(iig)或(iih)的化合物:其中代表与x2的连接点。[实施方案k]上面实施方案[a]、[b]或[j]中的任一个或根据本发明的其它实施方案的化合物,其中所述式(ii)的化合物是式(iia)的化合物。[实施方案l]上面实施方案[a]至[k]中的任一个或根据本发明的其它实施方案的化合物,其中x2是l1-l2-(l3)p-(l4)q-(l5)r;且l1选自:l2选自l3是其中r是氨基酸侧链;l4选自:l5是且p、q和r各自独立地是0或1,其中当p和q各自是0时,r必须是0。[实施方案m]上面实施方案[l]或根据本发明的其它实施方案的化合物,其中l3选自valcit、glyvalcit、valarg、phelys、alaala、glyglyphegly、alaalaala、alaasn、asnasn、asnala、valcitglypro、asnglypro、asnasnglypro、asn、glyasn、asnala、procitala、proasnleu、proasnala、propheala、prophegly、procitleu、proasnpro、proasnser和proasngly。[实施方案n]上面实施方案[l]或根据本发明的其它实施方案的化合物,其中:l1选自:l2是l3是valcit、glyvalcit、asnasn、asn或alaala;l4选自:l5是且p、q和r各自是0,或p、q和r各自是1。[实施方案o]上面实施方案[a]至[n]中的任一个或根据本发明的其它实施方案的化合物,其中x2通过ab的半胱氨酸残基、ab的赖氨酸残基或ab的谷氨酰胺残基、任选的谷氨酰胺295连接至ab。[实施方案p]上面实施方案[a]至[o]中的任一个或根据本发明的其它实施方案的化合物,其中ab是肿瘤靶向抗体、抗体片段、双特异性抗体或抗体片段、单克隆抗体、嵌合抗体或人源化抗体。[实施方案q]上面实施方案[a]至[p]中的任一个或根据本发明的其它实施方案的化合物,其中ab选自抗-her2抗体、抗-cd20抗体、抗-cd38抗体、抗-il-6受体抗体、抗-vegrf2抗体、抗-her-2抗体、抗-dll3抗体、抗-连接素4抗体、抗-cd33抗体、抗-cd79b抗体、抗-cd11a抗体、抗-bcma抗体、抗-cd22抗体、抗-trop2抗体、抗-frα抗体、抗-epcam抗体、抗-间皮素抗体、抗-liv1抗体、奥戈伏单抗、依决洛单抗、西妥昔单抗、针对玻璃体结合蛋白受体(αvβ3)的人源化单克隆抗体、阿仑珠单抗、用于治疗非霍奇金淋巴瘤的人源化抗-hla-dr抗体、131llym-1、用于治疗非霍奇金淋巴瘤的鼠抗-hla-drl0抗体、用于治疗霍奇金病或非霍奇金淋巴瘤的人源化抗-cd2 mab、拉贝珠单抗、贝伐珠单抗、替伊莫单抗、奥法木单抗、帕尼单抗、利妥昔单抗、托西莫单抗、伊匹木单抗、吉妥珠单抗、针对癌胚蛋白受体5t4的人源化单克隆抗体、m1/70(针对cd11b受体的抗体)、抗-mrc1、抗gcc、抗cd32和其它抗体。[实施方案r]式(iii)的化合物其中:r1选自c1-c10烷基、c1-c10氧杂烷基和c1-c10氮杂烷基;r2和r3各自独立地选自氢、c1-c5烷基和c1-c5烷氧基;n是1或2;y选自任选地被取代的芳基和任选地被取代的杂芳基;za选自-nrz-、-nrzc(o)-、-nrzc(o)-o-、-nrzc(o)-(ch2)k-nh-、-nrzc(o)-(ch2)k-o-、-nrzc(o)-o-(ch2)k-o-、-nrzc(o)-(ch2)k-n(ch3)-、-nrzc(o)-o-(ch2)k-nh-、-nrzc(o)-(ch2)k-nh-c(o)-o-和-nrzso2-;k是1至8的整数;rz选自氢、c1-c8烃、c1-c8氧杂烷基、c1-c8氮杂烷基、杂芳基和5-8元杂环;且xa选自氢、c1-c10烷基和-c(o)ch3,其中下述化合物被排除:[实施方案s]上面实施方案[r]或根据本发明的其它实施方案的化合物,其中:r1选自正丁基、-ch2oh和-ch2och2ch3;r2和r3各自是氢;n是1;y是苯基或吡啶基,其中每个是未被取代的或被卤素、c1-c4烷基、c1-c4烷氧基、c1-c4卤代烷基或c1-c4卤代烷氧基中的一种或多种取代;和rz当存在时是氢。[实施方案t]上面实施方案[r]或[s]中的任一个或根据本发明的其它实施方案的化合物,其中r1是正丁基且y是未被取代的苯基。[实施方案u]上面实施方案[r]至[t]中的任一个或根据本发明的其它实施方案的化合物,其中za选自-nrz-、-nrzc(o)-、-nrzc(o)-o-、-nrzc(o)-(ch2)k-nh-、-nrzc(o)-o-(ch2)k-nh-、-nrzc(o)-(ch2)k-nh-c(o)-o-和-nrzso2-。[实施方案v]上面实施方案[r]至[t]中的任一个或根据本发明的其它实施方案的化合物,其中za-xa选自-nhc(o)o(c1-c4)烷基、-nh2、-nhc(o)(ch2)knh2、-nhc(o)(ch2)knh-c(o)o(c1-c4)烷基和-nhc(o)(c1-c4)烷基。[实施方案w]上面实施方案[a]至[v]中的任一个或根据本发明的其它实施方案的化合物,其中k是1至4的整数。[实施方案x]上面实施方案[a]至[v]中的任一个或根据本发明的其它实施方案的化合物,其中k是1至6的整数,或k是1至3的整数,或k是1至2的整数,或k是2至4的整数,或k是2至3的整数。[实施方案y]一种药物组合物,其包含上面实施方案[a]至[x]中的任一个或根据本发明的其它实施方案的化合物、以及药学上可接受的载体、稀释剂或赋形剂。[实施方案z]上面实施方案[y]或根据本发明的其它实施方案的药物组合物,进一步包含治疗有效量的化学治疗剂。[实施方案aa]一种用于在受试者中刺激免疫应答的方法,所述方法包含在有效刺激免疫应答的条件下施用治疗有效量的上面实施方案[a]至[x]中的任一个或根据本发明的其它实施方案的化合物。[实施方案ab]上面实施方案[aa]或根据本发明的其它实施方案的方法,其中对具有癌症的受试者执行所述施用。[实施方案ac]上面实施方案[aa]或[ab]中的任一个或根据本发明的其它实施方案的方法,其中所述癌症是膀胱癌、乳癌、宫颈癌、结肠癌、子宫内膜癌、肾癌、肺癌、食管癌、卵巢癌、前列腺癌、胰腺癌、皮肤癌、胃癌、睾丸癌、胆管癌、结肠直肠癌、子宫内膜癌、头颈癌、甲状腺髓样癌、肾癌、眼癌、神经母细胞瘤、蕈样肉芽肿病、神经胶质和其它脑和脊髓肿瘤、肝癌、白血病、淋巴瘤或它们的任何组合。[实施方案ad]一种用于在受试者中诱导抗肿瘤免疫应答的方法,所述方法包含在有效诱导抗肿瘤免疫应答的条件下施用治疗有效量的上面实施方案[a]至[x]中的任一个或根据本发明的其它实施方案的化合物。[实施方案ae]上面实施方案[ad]或根据本发明的其它实施方案的方法,其中对具有肿瘤的选定受试者执行所述施用。[实施方案af]上面实施方案[ad]或[ae]中的任一个或根据本发明的其它实施方案的方法,其中所述肿瘤选自纤维肉瘤、粘液肉瘤、脂肉瘤、软骨肉瘤、成骨性肉瘤、脊索瘤、血管肉瘤、内皮肉瘤、淋巴管肉瘤、淋巴管内皮肉瘤、滑膜瘤、间皮瘤、尤因瘤、平滑肌肉瘤、横纹肌肉瘤、结肠癌、胰腺癌、乳癌、卵巢癌、前列腺癌、鳞状细胞癌、基底细胞癌、腺癌、汗腺癌、皮脂腺癌、乳头状癌、乳头状腺癌、囊腺癌、髓样癌、支气管原癌、肾细胞癌、肝细胞瘤、胆管癌、绒毛膜癌、精原细胞瘤、胚胎性癌、威尔曼瘤、宫颈癌、睾丸肿瘤、肺癌、小细胞肺癌、膀胱癌、上皮癌、神经胶质瘤、星形细胞瘤、髓母细胞瘤、颅咽管瘤、室管膜瘤、松果体瘤、血管母细胞瘤、听神经瘤、少突神经胶质瘤、脑膜瘤、黑素瘤、神经母细胞瘤和视网膜母细胞瘤。[实施方案ag]一种用于治疗肿瘤或异常细胞增殖的方法,所述方法包含在有效治疗肿瘤或异常细胞增殖的条件下施用治疗有效量的上面实施方案[a]至[x]中的任一个或根据本发明的其它实施方案的化合物。[实施方案ah]上面实施方案[ag]或根据本发明的其它实施方案的方法,其中所述肿瘤或异常细胞增殖是癌症。[实施方案ai]上面实施方案[ag]或[ah]中的任一个或根据本发明的其它实施方案的方法,其中所述癌症是膀胱癌、乳癌、宫颈癌、结肠癌、子宫内膜癌、肾癌、肺癌、食管癌、卵巢癌、前列腺癌、胰腺癌、皮肤癌、胃癌、睾丸癌、胆管癌、结肠直肠癌、子宫内膜癌、头颈癌、甲状腺髓样癌、肾癌、眼癌、神经母细胞瘤、蕈样肉芽肿病、神经胶质和其它脑和脊髓肿瘤、肝癌、白血病、淋巴瘤或它们的任何组合。[实施方案aj]一种用于治疗感染性疾病的方法,所述方法包含在有效治疗感染性疾病的条件下施用治疗有效量的上面实施方案[a]至[x]中的任一个或根据本发明的其它实施方案的化合物。[实施方案ak]上面实施方案[aj]或根据本发明的其它实施方案的方法,其中所述感染性疾病是病毒感染、细菌感染、真菌感染或它们的任何组合。[实施方案al]上面实施方案[aj]或[ak]中的任一个或根据本发明的其它实施方案的方法,其中所述感染性疾病是病毒感染。[实施方案am]上面实施方案[aj]至[al]中的任一个或根据本发明的其它实施方案的方法,其中所述感染性疾病选自冠状病毒、埃博拉、流感、肝炎、hib疾病、人免疫缺陷病毒(hiv)、人乳头瘤病毒(hpv)、脑膜炎球菌疾病、肺炎球菌疾病、麻疹、腮腺炎、诺如病毒、脊髓灰质炎、呼吸道合胞病毒(rsv)、轮状病毒、风疹病毒、带状疱疹、西尼罗河病毒、狂犬病病毒、肠道病毒、巨细胞病毒、疱疹病毒、水痘、黄热病、寨卡病毒或它们的任何组合。[实施方案an]上面实施方案[aj]或[ak]中的任一个或根据本发明的其它实施方案的方法,其中所述感染性疾病是细菌感染。[实施方案ao]上面实施方案[aj]或[ak]中的任一个或根据本发明的其它实施方案的方法,其中所述感染性疾病选自链球菌疾病、葡萄球菌疾病、白喉、脑膜炎球菌疾病、破伤风、百日咳、肺炎球菌疾病、细菌食品中毒、性传播的感染、结核病、莱姆病、肉毒中毒或它们的任何组合。[实施方案ap]上面实施方案[aj]或[ak]中的任一个或根据本发明的其它实施方案的方法,其中所述感染性疾病是真菌感染。[实施方案aq]上面实施方案[aj]或[ak]中的任一个或根据本发明的其它实施方案的方法,其中所述感染性疾病选自念珠菌病、组织胞浆菌病、皮肤癣菌病、足癣、曲霉菌病、隐球菌性脑膜炎、球孢子菌病或它们的任何组合。虽然在本文中已经描述和描绘了本发明的几个方面,但是本领域技术人员可以实现替代方面以实现相同目的。因此,所附权利要求旨在覆盖落入本发明的真实精神和范围内的所有这样的替代方面。
背景技术:
技术实现思路
- 还没有人留言评论。精彩留言会获得点赞!