组织释药浓度可控的抗癌药物递送系统及其制备方法与流程
1.本发明涉及医药领域,具体涉及组织释药浓度可控的抗癌药物递送系统及其制备方法。
背景技术:
2.迄今为止,化疗仍然是癌症治疗的主要手段之一。化疗之所以在癌症治疗中仍然有不可取代的地位,是因为化疗药物具有其他药物(如抗体类药物及细胞和基因治疗类药物)所不具备的功效:对癌细胞杀死能力强;抗癌谱广;在对癌细胞的杀灭作用和对正常细胞的损伤作用方面存在一定的浓度差异。在这些方面,目前并未有证据表明抗体类药物及细胞和基因治疗类药物比化疗药物做得更好。但在临床使用中,目前用于癌症治疗的药物制剂包括小分子化疗药物的普通制剂、抗体类药物、细胞治疗药物、基因治疗药物和各种纳米抗癌药物递送系统及其使用技术普遍存在以下问题:a.毒副作用强;b.癌瘤手术切除后,抗癌药物的作用目标减小或消失,在正常组织和癌组织中的分布比例增大或几乎完全分布于正常组织。因此,手术后使用抗癌药物主要或完全表现为毒副作用;c.抗癌药物需要在正常组织中持久存在,并在其中生成和维持有效的抗癌浓度,才能有效地避免细胞的癌变、癌细胞的转移和残留癌细胞形成癌灶。
3.d.由于a和b的原因,术后使用目前的抗癌药物,机体不能耐受其毒副作用而导致化疗失败。
4.e.从b和c可以看出,抗癌药物的作用强度增加,则毒副作用强度随之增加,因此治疗作用和机体耐受性之间存在着不可调和的矛盾:治疗作用要求药物作用强度增加,而机体而受性则要求药物作用强度降低。这是目前所有的抗癌药物所不能解决的问题。
5.f.解决这些问题的根本方法,是将药物(是所有的药物,而不仅仅是某一种具体的药物)制备成能够在正常组织中持久存在,既能有效地杀灭癌细胞,又不造成正常细胞明显损伤的药物制剂。但目前的制剂学并未有这种制备技术,因而这种对癌细胞致命,对正常细胞安全友好的药物更是无从谈起。
6.g.要制备对癌细胞致命,对正常细胞安全友好的药物,要有正确的吸附动力学理论,目前缺乏这种能够指导这种药物制备的吸附动力学理论。目前的吸附动力学理论只能对吸附实验的结果进行模拟,而不能用于对吸附实验过程的控制和吸附实验结果进行预测和判断。在缺乏这种能对实验过程进行控制和对实验结果进行预测和判断的理论的情况下,要制备出对癌细胞致命,对正常细胞友好安全的药物是不可能的。
7.例如,传统的一线抗癌药丝裂霉素c(mmc)对bgc-823胃癌细胞的最低有效浓度(ecmin)为1.1 mg/ml,而对淋巴细胞造成损伤的最小毒性浓度(tcmin)为40 mg/ml。很明显,我们只要将组织中的mmc浓度控制于1.1 mg/ml以上,40 mg/ml 以下,就能实现对癌细胞的选择性杀灭而不造成对淋巴细胞的损伤。科研实践证明,mmc在浓度为37 mg/ml,对
bgc-823癌细胞的杀灭率即大于95以上。这样,如果我们能将mmc在组织中的浓度控制于37 mg/ml,就可将癌细胞杀灭95%以上而对正常淋巴细胞不造成明显损伤。但是要正确使用对癌细胞致命,对正常细胞安全友好的药物,需要正确的释药动力学理论,目前缺乏这种释药动力学理论及对应的抗癌药物递送系统。
技术实现要素:
8.针对现有技术中的上述不足,本发明提供的一种组织释药浓度可控的抗癌药物递送系统及其制备方法解决了现有抗癌药物递送系统不能在正常组织中持久存在,无法长期保持对癌细胞进行抑制和杀灭作用而不对正常组织细胞造成明显损伤,难以兼顾对癌细胞致命、对正常细胞友好安全的问题。
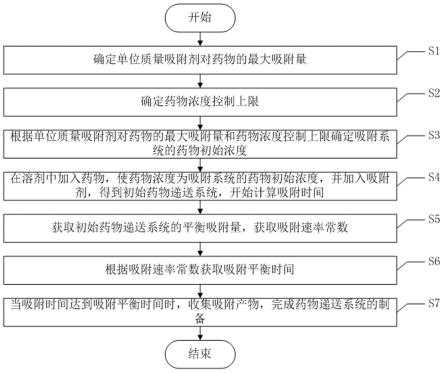
9.为了达到上述发明目的,本发明采用的技术方案为:提供一种组织释药浓度可控的抗癌药物递送系统的制备方法,其包括以下步骤:s1、确定单位质量吸附剂对药物的最大吸附量;s2、确定药物浓度控制上限;s3、根据单位质量吸附剂对药物的最大吸附量和药物浓度控制上限确定吸附系统的药物初始浓度;s4、在溶剂中加入药物,使药物浓度为吸附系统的药物初始浓度,并加入吸附剂,得到初始抗癌药物递送系统,开始计算吸附时间;s5、获取初始抗癌药物递送系统的平衡吸附量,获取吸附速率常数;s6、根据吸附速率常数获取吸附平衡时间;s7、当吸附时间达到吸附平衡时间时,收集吸附产物,完成抗癌药物递送系统的制备。
10.进一步地,步骤s1的具体方法为:采用药物浓度大于吸附浓度1000倍以上的吸附系统进行充分长时间吸附,收集吸附产物并测定吸附量,作为单位质量吸附剂对药物的最大吸附量。
11.进一步地,将药物对正常细胞的最低毒性浓度作为药物浓度控制上限。
12.进一步地,步骤s3的具体方法为:将药物浓度控制上限作为吸附平衡时的药物浓度,根据公式:获取吸附系统的药物初始浓度;其中为吸附剂浓度;为单位质量吸附剂对药物的最大吸附量。
13.进一步地,步骤s4的具体方法为:在体积为nv
l
毫升的溶剂中加入药物,使药物浓度为吸附系统的药物初始浓度,并加入吸附剂,使吸附剂浓度为,开始计算吸附时间,得到一个每v
l
毫升的溶剂中包含v
l
×
毫克的药物和v
l
×
毫克的吸附剂的初始抗癌药物递送系统;其中n为大于0的常数。
14.进一步地,步骤s5的具体方法为:
根据公式:获取初始抗癌药物递送系统的平衡吸附量;在吸附进行时间t时测定药物在溶剂中的浓度,并根据公式:获取吸附速率常数;其中表示以常数e为底数的对数。
15.进一步地,步骤s6的具体方法为:根据公式: 或 获取吸附平衡时间。
16.提供一种组织释药浓度可控的抗癌药物递送系统,其包括吸附剂和药物,并采用组织释药浓度可控的抗癌药物递送系统的制备方法制得。
17.本发明的有益效果为:1、本发明利用药物对癌细胞和正常细胞敏感性差异实现对癌细胞的选择性杀伤,由于制备的可控性,因此可以按照治疗需要制备出具有特定载药量、在组织中能够产生所需要的药物浓度、有效浓度范围、药物消除速率和有效浓度保持时间的抗癌药物递送系统,使抗癌药物递送系统在体内靶组织中的释药动力学精确可控。
18.2、本发明可以用于预防细胞癌变(抗癌变),预防癌细胞转移(抗转移)和残留癌细胞形成新的癌灶(抗复发)。
附图说明
19.图1为本制备方法的流程图;图2为实施例二和实施例三中acp-mmc ccdds有效浓度范围和acp-dox ccdds有效浓度范围图;图3为实施例四中不同治疗方式对实验性胃癌射频治疗后肿瘤复发的影响图;图4为实施例四中不同治疗方式对实验性胃癌射频治疗后复发肿瘤生长曲线的影响图;图5为实施例五中acp-mmc ccdds对人胃腺癌sgc-7901细胞移植癌的抗转移作用图;图6为实施例五中acp-mmc ccdds和mmc 对原位肿瘤的影响的比较图;图7为实施例六中acp-mmc ccdds 对hepg-2 细胞癌转移的抑制作用图;图8为实施例六中肝癌细胞腹腔种植后动物存活时间图;图9为实施例六中acp-mmc ccdds的组织和淋巴靶向性 figure 4 纳卡米妥腹腔注射后组织分布图;图10为实施例七中药物对h460 细胞胸腔种植模型的治疗作用活体成像观察图。
具体实施方式
20.下面对本发明的具体实施方式进行描述,以便于本技术领域的技术人员理解本发明,但应该清楚,本发明不限于具体实施方式的范围,对本技术领域的普通技术人员来讲,只要各种变化在所附的权利要求限定和确定的本发明的精神和范围内,这些变化是显而易见的,一切利用本发明构思的发明创造均在保护之列。
21.如图1所示,该组织释药浓度可控的抗癌药物递送系统的制备方法包括以下步骤:s1、确定单位质量吸附剂对药物的最大吸附量;s2、确定药物浓度控制上限;s3、根据单位质量吸附剂对药物的最大吸附量和药物浓度控制上限确定吸附系统的药物初始浓度;s4、在溶剂中加入药物,使药物浓度为吸附系统的药物初始浓度,并加入吸附剂,得到初始抗癌药物递送系统,开始计算吸附时间;s5、获取初始抗癌药物递送系统的平衡吸附量,获取吸附速率常数;s6、根据吸附速率常数获取吸附平衡时间;s7、当吸附时间达到吸附平衡时间时,收集吸附产物,完成抗癌药物递送系统的制备。
22.该组织释药浓度可控的抗癌药物递送系统,其包括载体和对靶向癌细胞敏感的抗癌药物,且:(1)在组织中释放的药物产生的药物浓度高于对癌细胞进行有效杀伤所需要的浓度而低于对正常组织细胞造成明显损伤的浓度;(2)有效浓度范围≥载体粒径的5倍;(3)有效浓度保持时间≥6个月;(4)载体是对抗癌药物具有可逆性吸附作用的吸附材料;(5)载体对靶组织正常细胞无毒或其毒性可以忽略;(6)载体在体内存在时间大于或等于有效浓度保持时间;(7)载体可以在体内代谢或从体内排出至体外;(8)载体的粒径为1~1000 nm,优选300~500nm;(9)载体材料是有机的或无机的;(10)采用组织释药浓度可控的抗癌药物递送系统的制备方法制得。
23.在具体实施过程中,组织释药浓度可控的抗癌药物递送系统的制备基于由发明人首次提出的“真”吸附动力学(tkt-rla)。tkt-rla中提出了“浓度选择性”用药理论:所有抗癌药物都具有治疗作用和毒副作用;治疗作用和毒副作用都与药物在体内环境中所产生的药物浓度有关;药物在靶组织中的治疗作用和毒副作用所需要的药物浓度不同;一般的抗癌药物的治疗作用所需要的浓度低于毒副作用所需要的浓度;将药物浓度控制于大于治疗浓度而低于毒副作用浓度,即可实现对癌细胞致命而对正常细胞安全,即可实现浓度选择性。药物在某一浓度水平时,可选择性杀伤癌细胞。这一水平的药物浓度称为药物的选择性浓度。依赖选择性浓度实现对癌细胞进行选择性杀伤的药物,称为浓度选择性药物。能够在靶组织中产生选择性药物浓度的抗癌药物递送系统,称为浓度选择型抗癌药物递送系统。本组织释药浓度可控的抗癌药物递送系统(ccdds)可在正常组织中持久存在而不造成正常细胞的明显损伤,因而可用于抗癌变、抗转移和抗复发。这是本ccdds与任何现有抗癌药物
的根本区别所在。
24.目前描述吸附平衡状态的动力学理论是langmuir方程和freundlich方程。按照这两个方程,吸附一定能够在有限时间内达到平衡状态,但由于其状态实质,便不能描述吸附动力学过程。而且,由于人们并不知道一个具体的吸附系统的达平衡时间,因而依靠实验所确定的“平衡”并不一定是平衡状态,因此不能准确模拟平衡状态的结果,模拟得到的吸附剂(载体)对吸附质(药物)的平衡吸附量与实际平衡吸附量只是在一定程度上一致,但差距很大。但tkt-rla认为:a.吸附一定可以在有限时间内达到平衡(这一点与langmuir方程和freundlich方程一致);b.系统中的吸附质按一级动力学降低,吸附剂对吸附质的吸附量按一级动力学增加(这一点与现有一级动力学一致);c.qe是系统组成成分与吸附剂和吸附剂相互作用性质(qm)的可解函数(这一点是本tkt-rla的原始创新)。
25.tkt-rla的基本方程包括:a、瞬时状态方程:其中v
l
为吸附系统溶剂的体积(注意:不是系统的总体积),c0和c
t
分别为吸附质的初始浓度和任一时刻的浓度(注意:tkt-rla中,浓度是系统中吸附质的量与溶剂体积v
l
的比值,而不是与系统总体积的比值);m为系统中吸附剂的质量;qm为单位质量吸附剂对吸附质的最大吸附量(反映吸附剂和吸附质相互作用的性质),与单位质量吸附剂对吸附质的吸附位点有关;e为自然对数的底;为吸附速率常数,也与吸附剂-吸附质相互作用的性质有关;t为吸附进行的时间。式1的文字描述为:溶剂(注意:不是溶液)中吸附质量的减少等于吸附剂吸附容量的减少,吸附剂的吸附容量按一级动力学速率减少。式1反映的是一个公理,但是当前的吸附动力学都不能正确的反映这一公理,因而不能得出正确的结果。langmuir方程和freundlich方程不能用于瞬时状态,因此现有的相关动力学方程得出的瞬时状态是错误的。
26.b、平衡吸附状态方程:用吸附平衡时的吸附质浓度ce取代式1中的c
t
,用吸附达平衡时间te取代式1中的t,即可得到平衡状态的基本方程:将式2与式1进行比较可知,平衡状态是吸附过程在一个特殊时间点的状态,因而te必定有确定值而不是
¥
,因而吸附必然在确定的时间达到平衡,这就从根本上颠覆了吸附在有限时间内不能达到平衡的概念。langmuir方程和freundlich方程与时间无关,因而无法用这两种方程得到达平衡时间。现有动力学的达平衡时间为
¥
,且与最大吸附量无关,因而是错误的。
27.c、静态平衡浓度ce公式:静态平衡浓度公式是指在不需要动力学参数,从吸附系统的组成和吸附剂的最大吸附量qm即可求出吸附质的平衡浓度ce的公式:
ꢀꢀꢀꢀꢀ
(3)
式3中的cm为吸附剂在溶剂中的浓度,是m与v
l
的比值。式3是tkt-rla的原始创新公式,与现有任何吸附动力学都无关,是使平衡状态方程完全可解的关键公式。在本ccdds制备中,将其作为一个经验公式使用即可。从式3可以看出,吸附平衡浓度ce不依赖于吸附过程而完全决定于系统的组成及其组成成分相互作用的性质。所有现有吸附动力学都未将反映吸附系统中各组分相互作用性质的qm考虑在内,因而不可能得到正确的结果。
28.d、吸附速率常数公式:观察式1和式2可以发现,吸附系统的一个状态中,v
l
、c0和m是可以人为控制的量,qm是可以用常规方法测得的量,e是常数,ce可以由式3计算得出,而和te都是需要测定的量,其中是可在吸附开始一定时间后即可测得的量。可从基本方程式1解出如式4。
29.ꢀꢀꢀꢀꢀꢀ
(4)式4中的t完全是人为决定的测定时间,c
t
是在吸附开始后t时刻测得的溶剂中吸附质浓度。是使吸附过程完全可解的关键参数。由一个时间点得出的可预计出任何时间点的吸附质浓度c
t
。langmuir方程和freundlich方程与时间无关,因而无法用这两种方程得到。现有动力学与最大吸附量无关,得出的值是错误的。用平衡浓度ce取代式4中的瞬时浓度c
t
,用吸附达平衡时间te取代时刻t,即可得到由平衡状态计算的公式5。(公式5也可以由基本方程式2解出)。
30.ꢀꢀꢀꢀꢀꢀꢀꢀ
(5)式5将反映平衡状态的ce与反映吸附过程的吸附速率常数联系起来,解决了迄今为止的所有的吸附动力学理论所不能解决的一个难题:吸附过程方程与吸附状态方程不能得到一致的结果。
31.e、吸附达平衡时间te公式:在得到和ce值后,将其代入基本方程式2,te可以容易地解出如式6。
32.ꢀꢀꢀꢀꢀ
(6)式6表明吸附达平衡时间te是系统组成和吸附速率的函数。如果和c
e 已知,te也可用式7由和ce值求得。
33.ꢀꢀꢀꢀꢀꢀ
(7)式7和式6的一致性在理论上容易证明。式7表明吸附达平衡时间te是吸附速率和吸附质初始与平衡状态浓度的函数。式6和式7共同表明吸附达平衡时间不仅是存在的,而且是精确可求的。能够求出te是tkt-rla的独特能力,目前所有的吸附动力学理论都不能做到这一点。
34.f、吸附质瞬时浓度公式:从基本方程式1,可解出任一时刻的吸附质浓度c
t
公式如式8。
35.ꢀꢀꢀꢀꢀ
(8)测得后,即可用式8求得任一时刻吸附质的浓度c
t
。
36.g、吸附质平衡浓度公式:从基本方程式2,可解出吸附平衡时的吸附质浓度ce公式如式9。式9也可以ce取代式8中的c
t
,以te取代式8中的t而直接得到。
37.ꢀꢀꢀꢀꢀ
(9)h、任一时刻时的单位质量吸附剂对吸附质的吸附量公式:tkt-rla基本方程(式1)的两边都是某一时刻系统中吸附剂总量所已经吸附的吸附质总量。吸附质的总量与吸附剂质量的比值,即是某一时刻的单位质量吸附量q
t
。用式1的左边除以吸附剂质量m并经化简可得到q
t
公式10。
38.ꢀꢀꢀꢀꢀꢀꢀ
(10)用式1的右边除以吸附剂质量m可得到q
t
公式11。
39.ꢀꢀꢀꢀꢀ
(11)观察公式10和11可以发现在公式10中不含有反映吸附性质的参数qm、时间t和吸附速率,因此它是一个状态公式,它反映的是一个公理,其中的c
t
可由实验测得;公式11含有qm、a和t,因此它是一个动态公式,它反映的是吸附动力学过程,其中qm可由常规实验测定,可以通过公式4由c
t
计算,t则可以人为地决定。
40.i、吸附平衡时的单位质量吸附剂对吸附质的吸附量公式:用式2的左边除以吸附剂质量m并经化简可得到qe公式12。
41.ꢀꢀꢀꢀꢀꢀꢀ
(12)用式2的右边除以吸附剂质量m可得到qe公式13。
42.ꢀꢀꢀꢀꢀ
(13)综上所述,式1~13形成了用tkt-rla计算吸附系统量的方面的各种参数的完整的方程和公式系统,基于tkt-rla,即可实现本组织释药浓度可控的抗癌药物递送系统。具体制备过程如下:a、确定单位质量吸附剂对药物的最大吸附量:用药物浓度比吸附剂浓度大1000倍以上的吸附系统,吸附进行充分长时间,直到吸附量不再发生变化,收集吸附产物,测定其吸附量,这一吸附量可视为吸附剂对药物的最大吸附量qm。
43.b、确定浓度控制范围:将靶组织正常细胞和治疗对象癌细胞分别进行细胞培养,在培养液中加入不同浓度的药物,测定对正常细胞的最低毒性浓度(c
tn
)和对癌细胞的最低毒性浓度(c
tc
)。以c
tn
和c
tc
分别作为浓度控制的上限(ch)和下限(c
l
)。由于半数毒性浓度(c
tn50
)比较容易测定,对于一些生理上不太重要组织,可将c
tn50
作为浓度控制的上限ch。
44.c、确定浓度控制水平:在浓度控制范围中选择浓度选择药物浓度控制水平(cc)。为提高治疗效果,cc越高越好,因此cc一般选择为接近ch但低于ch。为了便于取值,cc可以直接取值为ch。
45.d、确定吸附系统吸附质初始浓度c0:将ch作为吸附平衡时的吸附质浓度ce,确定c0。由tkt-rla的平衡浓度公式(式3)可解出初始浓度公式如式14。
46.ꢀꢀꢀꢀꢀꢀ
(14)式14中的载体浓度cm和溶剂体积(v
l
)可依据便于收集产物的原则人为地给定。
47.e、确定系统组成:在体积为v
l
的溶剂(依药物溶剂而异,水溶性药物使用纯水。在不同溶剂中,平衡状态浓度、最大吸附量不同,必须各自测定)中加入药物,使药物浓度为c0;加入载体,使载体浓度为cm。如此便组成一个由v
l ml溶剂,含v
l
×c0 mg的药物和含v
l
×cm mg载体的吸附系统。
48.f、确定平衡吸附量:在平衡浓度ce和各组分确定后,平衡吸附量qe便可用式12确定。
49.g、吸附:使吸附系统在给定环境条件下进行吸附。方便起见,吸附可在室温下静置进行。为节省时间,吸附可将系统置于超声场中进行。吸附的环境条件可影响吸附速率和达平衡时间,但不影响平衡吸附浓度和平衡吸附量。
50.h、测定吸附速率常数:在吸附进行一段时间t时测定药物在溶剂中的浓度c
t
,即可用式4计算吸附速率常数。需注意t《te,否则测出的速率常数是错误的,会得到偏小的和偏大的te,t比te大得越多,偏差越大。确保t《te的办法是进行预试验。预试验是在2个时间点测定2次c
t
。设在时间t1测到c
t1
,在时间t2测到c
t2
。如果c
t1
》c
t2
,则可以肯定t1《te;如果c
t1
=c
t2
,则两个时间点都大于te,即t2》t1》te必须在小于t1的范围内重新寻找小于te的时间点。
51.i、确定吸附达平衡时间:得到值后,即可用公式6或7计算达平衡时间te。
52.j、吸附继续进行至达平衡时间之后,收集吸附产物,即可得到载药量为qe的ccdds。
53.实施例一:以制备浓度控制型淋巴靶向性抗癌药物递送系统(靶向淋巴系统和胃癌和肝癌的活性炭-丝裂霉素ccdds(acp-mmc ccdds)的制备)为例:(1) 吸附系统设计:a实验测定,丝裂霉素c(mmc)对淋巴细胞的最小毒性浓度为40μg/ml,对胃癌细胞进行杀灭的最小有效浓度为1.1μg/ml。因此,acp-mmc ccdds在组织液中的平衡浓度设为37μg/ml。b实验测定,300 nm活性炭(acp)对mmc的最大吸附量为qm=280μg/mg。ccdds制备系统acp浓度设为cm=175μg/ml。c用rla-tk初始浓度c0公式确定acp-mmc吸附系统中mmc的初始浓度为c0=0.06492467 mg/ml≈65μg/ml。
54.(2) 用rla-tk确定在mmc初始浓度c0=64.92μg/ml和acp浓度cm=175μg/ml 的吸附系统中acp对mmc的平衡吸附量qe为0.1595695438 g/g≈160 mg/g。qe可用两种方式计算:mg/g。qe可用两种方式计算:(3) 吸附:在三蒸水中加入mmc,使其初始浓度为c0=65μg/ml;加入acp使其浓度为cm=175μg/ml;25 ℃条件下进行吸附。
55.(4) 吸附速率常数:在吸附进行30 min时测得溶液中mmc浓度为c
30
=48.65μg/ml,由tk-rla的速率常数公式计算出=2.255
×
10-4
/s。
56.(5)用rla-tk的达平衡时间te公式由吸附速率常数、初始浓度c0、活性炭浓度cm和活性炭对mmc的最大吸附量qm=280 mg/g计算吸附达平衡时间为3744 s=1.04 h。
57.(6) 吸附继续进行至1.04 h后,即可得到平衡浓度ce=37μg/ml,载体载药量为qe=160 mg/g,制剂含药量为13.79%的acp-mmc ccdds。
58.(7) 有效浓度范围:在tk-r的距粒子中心nr处的浓度公式中,令cn为对癌细胞的最低有效浓度,则可从中解出有效浓度范围re1=5.8 r。如图2所示(图2中re1指向的实线圆的半径为re1;re2指向的虚线圆的半径为re2),因为r=300 nm,因此以距粒子中心距离表示的有效浓度范围re1=5.8
×
0.15=0.87μm。
59.(8)acp-mmc ccdds在腹膜腔隙组织液中的有效浓度保持时间(t
ecm
):腹腔注射的acp-mmc ccdds主要存在于壁层腹膜和脏层腹膜之间腹膜间隙和淋巴组织中。腹膜的厚度为1~2 mm,取平均值1.5 mm。毛细淋巴管直径为30~80μm,取平均值55μm则半径r=27.5μm。用cn公式计算一个贴近腹膜的acp-mmc ccdds粒子在腹膜外缘处的药物浓度。淋巴流体中心距acp-mmc ccdds粒子中心的距离与粒子半径r的比值n=10183。acp-mmc ccdds在淋巴流体中心产生的药物浓度c
10183
=3.5682
×
10-7
μg/ml。毛细淋巴管中淋巴液流量为3
×
10-7
μl/s=3
×
10-10 ml/s。流体淋巴液对药物的消除速率为qf=1.07046
ꢀ×
10-16
μg/s。药物的消除速率常数为k=9.64
×
10-9
/s。有效浓度保持时间tecm=3.647
×
10
8 s=1.013
×
10
5 h=4.221
×
10
3 d=11.56 y。浓度半衰期t
1/2
=7.190
×
10
7 s=1.997
×
10
4 h=832 d=2.279 y。淋巴小结的大小尺度与腹膜厚度近似,因此与腹膜腔隙中的acp-mmc ccdds相似的有效浓度保持时间tecm和半衰期t
1/2
。
[0060]060]060]060]060]060]
(9) acp载药量与有效浓度保持时间之间的关系:acp-mmc ccdds的有效浓度tecm不仅与其浓度消除速率常数有关,而且与其载药量有关。实验测得,acp的比重dc=0.5 g/cm3。一个acp粒子直径为300 nm,半径r
p
=150 nm=150
×
10-7
=1.5
×
10-5 cm,体积v
p
=1.413
ꢀ×
10-14 ml,重量wc=1.76625
×
10-15 g。acp的单位质量载药量qm=160 mg/g。因此, acp的粒子载药量为q
p
=2.826
×
10-16 g/p=2.826
×
10-10
μg/p。因此,以粒子体积计算的药物浓度为c
p
=20 mg/ml。粒子表面浓度即药物在水溶液中的浓度为cs=37μg/ml。二者之比为20000/37=540.54。这一浓度比足以使cs得到长时间的维持。毛细淋巴管淋巴液流量f=3
×
10-10 ml/s,某时刻淋巴流体中心初始药物浓度由式1算得,t=0时,为c
f0
=3.56821
×
10-7
μg/ml;t=30 min=1800 s时,c
f30
=3.56815
×
10-7
μg/ml;在t=t
ecm
=3.647
×
10
8 s时,c
ftecm
= 1.0607
×
10-8
。
ccdds制备系统acp浓度设为cm=250μg/ml;c用rla-tk确定acp-dox ccdds制备系统中dox初始浓度c0为0.107 mg/ml=107μg/ml。
[0064]
(2) 用rla-tk确定在dox c0=107μg/ml和cm=250μg/ml 的吸附系统中acp对dox的平衡吸附量qe为0.148 g/g≈148 mg/g。qe可以用c0、ce和cm进行计算,也可以直接用tk-r的静态平衡吸附量公式计算。
[0065]065]
(3) 吸附:在三蒸水中加入dox,使其初始浓度为c0=107μg/ml;加入acp使其浓度为cm=250μg/ml;25 ℃条件下进行吸附。
[0066]
(4) 测定acp对dox的吸附速率常数:在吸附进行30 min时测得c
30
=85.72μg/ml,由tk-rla的吸附速率常数公式计算出=2.6257
×
10-4
/s。
[0067]
(5) 用rla-tk的达平衡时间公式由吸附速率常数、初始浓度c0、活性炭浓度cm和活性炭对dox的最大吸附量qm=280 mg/g确定吸附达平衡时间为4046 s=1.124 h。
[0068]
(6) 继续吸附至1.124 h,即可得到平衡浓度ce=70 μg/ml,载体载药量为qe=148 mg/g,制剂含药量为12.9%的acp-dox ccdds。
[0069]
(7) 有效浓度范围:在tk-r的距粒子中心nr处的浓度公式中,令cn为对癌细胞的最低有效浓度(2 μg/ml),则可从中解出n=5.916,有效浓度范围re2=5.916 r。如图2所示,因为r=300 nm,因此以距粒子中心距离表示的有效浓度范围re2=5.916
×
0.15=0.8874μm。
[0070]
(8) acp-dox ccdds在胸膜腔隙组织液中的有效浓度保持时间(tecm):胸腔注射的acp-dox ccdds主要存在于壁层胸膜和脏层胸膜之间胸膜间隙和淋巴组织中。胸膜的厚度与腹膜的厚度相等,因而胸膜腔中acp-dox ccdds药物的消除速率常数与acp-mmc ccdds在腹膜腔隙中的消除速率常数相等,为k=9.64
×
10-9
。胸膜腔中acp-dox ccdds药物有效浓度保持时间为tecm=3.688
×
10
8 s=1.024
×
10
5 h=4.268
×
10
3 d=11.69 y。胸膜腔中acp-dox ccdds药物浓度半衰期与acp-mmc ccdds在腹膜腔隙中的浓度半衰期相等,为t
1/2
=7.190
×
10
7 s=1.997
×
10
4 h=832 d=2.279 y。淋巴小结的大小尺度与腹膜厚度近似,因此与腹膜腔隙中的ccdds相似的有效浓度保持时间tecm和半衰期t
1/2
。
[0071]
(9) 制剂形式:由本实施例得到的acp-dox ccdds也可有二种制剂形式。其一为粉针剂,是活性炭与mmc质量比为2.34:1的混合物,使用前加入适量三蒸水使其mmc浓度为c0=
107μg/ml,acp浓度为cm=250μg/ml。在超声场中静置1 h后使用。根据临床dox静脉注射用量为262.5 mg,因此活性炭的用量应为614.25 mg。因此,临床制剂可为876.75 mg/支,使用前加水2453 ml,在超声场中静置 1 h后胸腔滴入。其二为液体针剂,是将262.5 mg dox,614.25 mg acp 和2453 ml水装入一个输液袋。临床上可直接胸腔输入或滴入。
[0072]
本例制备的acp-dox ccdds,主要用于靶向淋巴系统,预防肺组织细胞的癌变和肺癌术后的复发。给药方式主要为胸腔输入、滴入和术区注射但不限于胸腔和术区注射。
[0073]
实施例三:用acp-mmc ccdds腹腔注射治疗实验性胃癌:(1)动物胃癌模型:取处于对数生长期的体外培养的 低分化人胃癌bgc-823 细胞,胰酶消化后吹打成细胞悬液,1000 rpm,离心 5 min,弃去培养液,用无菌生理盐水重悬细胞制备单细胞悬液,并调细胞浓度至 10
7 个/ml。以每只 0.2 ml 注射至 4 只 babl/c-nu 裸鼠腹部皮下。当皮下移植瘤长至直径为1 cm 左右时取出移植瘤,剔除周围纤维包膜,将瘤组织切成约 l mm
3 的小块,直接切开裸鼠右下皮肤植入,2 只/ 代做鼠间传代。取稳定传代 3 代裸鼠皮下移植瘤。去除纤维包模及坏死组织,并切成 1 mm
3 的瘤块,种植于右下腹部。
[0074]
(2)药物治疗:制备的模型动物48 只,雌雄各半,18 ~ 22 g,随机平均分为8组:生理盐水对照1组,空白acp (5 mg/kg,为含有1 mg/kg mmc的载体剂量)对照1组,mmc 0.33、1和 3 mg/kg 3 个剂量组,ccdds(以其所含的mmc量计)0.33、1 和 3 mg/kg 3 个剂量组。另取用于活体成像36只16~20 g babl/c-nu裸鼠,雌雄各半,平均分为6组:生理盐水对照 1 组,空白acp 对照 1 组, mmc 1 mg/kg 组和ccdds(以其所含的mmc 剂量计)0.33、1 和 3 mg/kg 3 个剂量组。瘤细胞接种 1 周后称重,按上述剂量腹腔一次性给药。右侧腋窝种植模型按上述剂量瘤周淋巴引流区皮下一次性注射。对照组给予等体积生理盐水以药物组相同的途径注射。
[0075]
(3)治疗效果:a对肿瘤体积的抑制:肿瘤体积变化如表1。用组织块移植1周后 肿瘤大小平均 0.42
±
0.9 cm3。此时由于尚未用药,各组肿瘤体积无明显差异。对照组肿瘤增长迅速,接种后的第二周增长2.3倍;第三周时达第一周的3.6倍;第四周时达第一周的5.1倍。空白载体acp 对肿瘤的生长没有抑制作用。mmc 和ccdds都能剂量依赖性地明显抑制肿瘤的增长,但在相同剂量下,ccdds的作用较mmc 明显为强,且随给药后时间的延长,ccdds对肿瘤的抑制作用较 mmc 更进一步地增强。于给药后第一周,ccdds作用较mmc 有增强的倾向,但不具有统计学意义。于给药后第二周,ccdds作用与 mmc 相比明显增强,具有统计学意义。于给药后第三周,ccdds作用更明显地强于mmc。在 3 mg/kg 剂量下,给药三周后ccdds的抑瘤作用近于mmc 的2 倍。如表2所示,动物死亡后及第21 天动物处死后剥出肿瘤的瘤重与体积测量的结果一致。在0.33 ~ 3.00 mg/kg 范围内,各剂量组ccdds抑瘤作用均比等剂量下的 mmc 为强。在同为3 mg/kg 剂量下,ccdds 的抑瘤率为 46.2%,而cbmc的抑瘤率达69.4%。
[0076]
(4)结论:acp-mmc ccdds对实验性胃癌具有良好的治疗作用。
[0077]
表1:胃癌bgc-823实体型裸鼠腹腔种植模型肿瘤体积变化
表2:药物对胃癌bgc-823实体型裸鼠移植模型瘤重的影响表1中mean
±
sd, n=6。腹腔注射给药。mmc:丝裂霉素;cbmc:acp-mmc ccdds的一种制剂。* p 《 0.05, ** p 《 0.01, *** p 《 0.001 与生理盐水相比;+ p 《 0.05,++ p 《 0.01,+++ p 《 0.001,与相同剂量的 mmc 相比。
[0078]
表2中mean
±
sd,n=6。腹腔注射给药。mmc:丝裂霉素;cbmc:acp-mmc ccdds的一种制剂。* p 《 0.05, ** p 《 0.01,*** p 《 0.001 与生理盐水相比;+ p 《 0.05, ++ p 《 0.01,+++ p 《 0.001,与相同剂量的 mmc 相比。
[0079]
实施例四:用acp-mmc ccdds射频灶注射治疗实验性胃癌(1)动物胃癌模型:取处于对数生长期的体外培养的 低分化人胃癌bgc-823 细胞,胰酶消化后吹打成细胞悬液,1000 rpm,离心 5 min,弃去培养液,用无菌生理盐水重悬细胞制备单细胞悬液,并调细胞浓度至 10
7 个/ml。以每只 0.2 ml 注射至 4 只 babl/c-nu 裸鼠腹部皮下。当皮下移植瘤长至直径为1 cm 左右时取出移植瘤,剔除周围纤维包膜,将瘤组织切成约 l mm
3 的小块,直接切开裸鼠右下腹皮肤植入,2 只/ 代做鼠间传代。取稳定传代 3 代裸鼠皮下移植瘤。去除纤维包模及坏死组织,并切成 1 mm
3 的瘤块,种植于右侧腋下。
[0080]
(2)射频治疗:当移植瘤长至 1.5 cm时给予射频(rfa)治疗。 动物用 0.004 ml/g 10% 水合氯醛麻醉。小鼠固定于电极板,确保电极板,小鼠和射频电极之间无绝缘物质。瘤区和射频针用75%乙醇消毒。射频针垂直插入肿瘤中心,射频频率500 khz,功率 5w,温度上限70℃,开通电源,温度达到60℃开始计时,射频照射2 min。照射后小鼠放回鼠笼,保持温暖至清醒。
[0081]
(3)射频灶药物注射:mmc 溶于生理盐水,终浓度为1 mg/ml。 acp和ccdds 分别混悬于生理盐水,终浓度分别为 5 mg/ml 和 6 mg/ml。以1 mg/kg mmc的剂量,用微量注射器沿射频电极路线和深度,在射频治疗后立即一次注射入射频灶内。acp和ccdds悬液在注射前振摇。
[0082]
(4)射频后肿瘤复发的监测:在射频治疗后第1,3,9和15天,用游标卡尺测量复发肿瘤的长度和宽度。肿瘤体积用式v (ml) = a
×
b2/2 (ml) 计算。各组间肿瘤大小以肿瘤体积为准进行比较。肿瘤抑制率由式ir=(治疗前肿瘤体积-治疗后肿瘤体积/治疗前肿瘤体积)
×
100%计算。抗瘤率用ar (%)= (对照组肿瘤体积
ꢀ–
治疗组肿瘤体积)/ 对照组肿瘤体积
×
100%。
[0083]
(5)治疗结果:典型的射频治疗后肿瘤的复发情况对比于图3。所有射频治疗后的胃癌均有明显的复发,单用acp和单用mmc均不能阻止复发,但用acp-ccdds治疗组有60%小鼠基本没有癌瘤复发。如图4所示,其余小鼠有复发,复发的癌瘤小于对照组及单用acp。mmc和ccdds配合射频治疗组均可抑制复发癌瘤的生长(p《0.05),但这两种治疗方式之间无统计学上的显著性差别(p》0.05)。
[0084]
(6)结果分析:mms+raf和ccdds+raf两种治疗方式在抑制复发癌瘤方面无显著差别,表明ccdds的主要治疗效果在于ccdds可以使射频治疗后的胃癌不能复发。一旦复发发生,ccdds和mmc对复发癌瘤生长的影响,差别不大,虽然ccdds的抑制作用有大于mmc的倾向。因为动物实验受实验周期的限制,因此在有限时间内,ccdds的长期效应不能得到充分发挥。癌瘤复发是射频治疗后的即时现象,因此ccdds对复发的影响可以在本实施例中容易地观察到。
[0085]
(7)实验结论:acp-mmc ccdds可以阻止射频治疗后的癌复发,因此acp-mmc ccdds配合射频治疗,具有使癌得到根治的潜力。
[0086]
实施例五:用acp-mmc ccdds对人胃腺癌sgc-7901细胞移植癌的抗转移作用:(1)动物模型:采用人未分化胃癌sgc-7901 细胞株对babl/c-nu裸小鼠进行腹腔种植。取处于对数生长期的体外培养的sgc-7901细胞,胰酶消化后吹打成细胞悬液,1000 rpm,离心5 min,弃去培养液,用dmem 培养基重悬细胞制备单细胞悬液,并调细胞浓度至107个/ml。以每只0.2 ml注射至babl/c-nu裸小鼠的腹部皮下。当皮下移植瘤长至直径为1 cm 左右时取出移植瘤,剔除周围纤维包膜,将瘤组织切成约 l mm
3 的小块,直接切开裸鼠腋下皮肤植入,2 只/ 代做鼠间传代。取稳定传代 3 代裸鼠皮下移植瘤。去除纤维包模及坏死组织,并切成 1 mm
3 的瘤块,种植于右下腹部皮下,作为实验模型。
[0087]
(2)药物治疗:制备的模型动物48 只,雌雄各半,18 ~ 22 g,平均分为8组:生理盐水对照1组,空白acp (5 mg/kg, 为含有1 mg/kg mmc的载体剂量)对照1组,mmc 0.33、1和 3 mg/kg 3 个剂量组, acp-mmc ccdds(以其所含的mmc 剂量计)0.33、1 和 3 mg/kg 3 个
剂量组。瘤块接种后随机分组按上述剂量一次性腹腔注射。对照组给予0.2 ml/只生理盐水腹腔注射。空白acp 对照组给予未载药的acp 5 mg/kg 腹腔注射。
[0088]
(3)肿瘤检测:a对原位肿瘤的抑制作用:对死亡小鼠,解剖大体观察,剥取原位肿瘤称重,记录,待小鼠全部死亡后,对瘤重进行统计分析,按公式:瘤重抑制率 = (对照组瘤重—用药组瘤重)/对照组瘤重
´
100 计算对原位肿瘤的抑制率。b 对转移的抑制作用:除对原位肿瘤剥瘤称重,同时观察肿瘤的转移情况,计数转移灶数,并以肿瘤转移灶数作为转移评价的指标。按公式:转移抑制率 = (对照组转移灶数—用药组转移灶数)/对照组转移灶数
´
100% 计算转移抑制率。
[0089]
(4)实验结果:转移情况如图5所示:人未分化胃癌sgc-7901细胞是常用的胃癌转移模型的细胞系。因此,除了在原位发生肿瘤,在此模型上发现有广泛的腹腔转移,六只小鼠均有不同程度的转移发生。转移灶大小不等。大者可接近移植原位癌的大小,小者只有约 1 mm。转移癌在形态上不同于原位癌。原位癌呈椭圆形,颜色暗红,表面光滑。转移癌形态多样,可呈分叶状、结节状,表面凸凹不平。颜色可为白色或灰色。转移部位以肠壁最多,其次为肝下。转移灶数最少者有10 个,最多者达32 个。如表3所示,mmc 对转移灶数无明显影响,而acp-mmc ccdds可明显减少sgc-7901 细胞癌转移的发生率,尤其是在所用高剂量 3 mg/kg 情况下,6 只小鼠均未见有转移发生。
[0090]
b 原位肿瘤如图6所示:acp-mmc ccdds能抑制未分化胃癌sgc-7901细胞在腹腔种植部位所形成的原位肿瘤的生长,使肿瘤明显减小,瘤重明显减轻,如表4所示。mmc和acp-mmc ccdds对瘤重都有抑制作用,但acp-mmc ccdds较mmc 作用更强。在剂量为 0.33 mg/kg 时,mmc 对瘤重的抑制率只有约 12%,没有统计学意义,但acp-mmc ccdds已具有明显的抑制作用,可达50%以上,具有明显的统计学显著性。在剂量为 1 mg/kg 时,mmc 对瘤重的抑制率可达约25%,具有统计学意义,但acp-mmc ccdds在此剂量下对瘤重的抑制率已达65%。acp-mmc ccdds的抑制率明显高于 mmc,两组相比有极显著的统计学意义。在实验所用的最高剂量 3 mg/kg 时,mmc对瘤重的抑制率为38.2%,而acp-mmc ccdds在此剂量下可达87.5%。尤其值得注意的是在此剂量下,有 3 只小鼠未观察到有肿瘤的发生。
[0091]
(5)结论:acp-mmc ccdds明显抑制胃癌转移,在充分剂量下可完全避免转移;acp-mmc ccdds明显抑制原位肿瘤的生长,充分剂量下可使原位肿瘤完全消退。这些结果提示acp-mmc ccdds具有使胃癌得到根治的潜力。
[0092]
表3:药物对 sgc-7901细胞裸鼠移植癌转移灶数的影响
表4:药物对未分化胃癌sgc-7901细胞瘤重的影响表3中mean
±
sd,n = 6。腹腔注射给药。mmc:丝裂霉素;cbmc:acp-mmc ccdds的一种制剂。 ***
表示与对照组相比,p﹤0.001;
△△
表示与mmc组相比,p﹤0.01;
△△△
表示与mmc组相比,p﹤0.001。发生率的比较用检验,转移率的比较用t 检验。
[0093]
表4中mean
±
sd,n = 6。腹腔注射给药。mmc:丝裂霉素;cbmc:acp-mmc ccdds的一种制剂。
* p 《0.05,***,p﹤0.001与对照组相比;
△△△
p﹤0.001与mmc 组相比。
[0094]
实施例六:acp-mmc ccdds对人hepg-2肝癌的抗复发作用:(1)动物模型:采用人hepg-2肝癌 细胞株对babl/c-nu裸小鼠进行腹腔种植。取处于对数生长期的体外培养的hepg-2肝癌细胞,胰酶消化后吹打成细胞悬液,1000 rpm,离心5 min,弃去培养液,用dmem 培养基重悬细胞制备单细胞悬液,并调细胞浓度至108个/ml。以每只0.4 ml注射至babl/c-nu裸小鼠腹腔,模拟肝癌手术后腹腔种植转移造成的复发。于移植后第四周剖腹观察肿瘤形成情况,取腹腔组织及其引流淋巴结做病理组织学观察。如动物死亡,则于死亡时进行剖腹观察和取材。剖腹观察和病理组织学取材后缝合腹腔,继续观察动物死亡情况。
[0095]
(2)药物治疗:制备的模型动物36 只,雌雄各半,18 ~ 22 g,平均分为4组:生理盐水对照和空白acp (5 mg/kg,为含有1 mg/kg mmc的载体剂量)对照各1组,mmc 3 mg/kg 2组分别在肿瘤细胞种植前1天和30天腹腔一次性腹腔注射。acp-mmc ccdds(以其所含的mmc 剂量计)3 mg/kg 2组,分别在肿瘤种植前1天和30天腹腔一次性腹腔注射。
[0096]
(3)实验结果:a 对肝癌腹水的影响:hepg-2 肝癌细胞腹腔种植可引起腹水形成。生理盐水和空白对照小鼠均有不同程度的腹水形成。mmc于种植前前1天和30天给药没有预防腹水形成的作用。acp-mmc ccdds种植前前1天和30天给药动物均未见有明显的腹水形成。
[0097]
b 对肿瘤复发的影响。如图7所示,hepg-2 肝癌细胞腹腔种植模拟了腹腔种植转移引起的复发,可腹腔形成大小不等的结节,经病理组织学观察为癌组织。转移复发灶最多见于肠壁,其次为肝下、胃周、大网膜、肠系膜等。肉眼可见转移灶最少者为34 个,最多者达56 个。像在对sgc-7901 细胞转移癌的情况一样,mmc 对hepg-2 肝癌的转移没有明显的抑制作用,各剂量组与对照相比没有统计学意义,也没有剂量依赖性。如表5所示,acp-mmc ccdds可以剂量依赖性地抑制hepg-2 细胞癌的种植复发。在剂量为 0.33 ~ 1.00 mg/kg,acp-mmc ccdds虽然对癌转移的以小鼠只数为单位计算的发生率没有明显的影响(没有统计学显著性),但转移灶数都有明显的减少,具有统计学显著性。在实验所用的最大剂量 3 mg/kg 剂量下,acp-mmc ccdds治疗组 6 只小鼠均未观察到有转移的发生,癌转移发生率与对照组比较有统计学意义(p 《 0.05,x
2 检验)。转移灶数减少则更为明显。在低剂量组,对癌灶数量的减少与对照组相比已统计学显著性;在中高剂量组,cbmc对癌转移灶数的减少与相应的 mmc 治疗组相比均有统计学意义。
[0098]
c对死亡时间的影响:空白和acp对照组动物均于癌细胞种植后四周内死亡。mmc于种植前前1天和30天给药没有延长存活时间的作用,动物与空白和acp对照组一样在癌细胞种植后四周内全部死亡。如图8所示,acp-mmc ccdds种植前1天和30天给药则明显延长动物存活时间。acp-mmc ccdds种植前1天给药组的六只动物存活时间均大于10个月,13个月后仍有三只存活。acp-mmc ccdds种植前30天给药组的六只动物存活时间均大于8个月,13个月后仍有四只存活。这说明前30天给药与前1天给药几乎具有同样的预防复发作用。这种使动物长期存活的作用是由于acp-mmc ccdds使腹腔组织mmc长期维持其对癌细胞的有效杀伤浓度,但又不对正常组织细胞造成明显损伤的结果。mmc不能延长动物的存活时间,是因为mmc腹腔注射后被迅速排泄,有效浓度维持时间短,在其注射后的高浓度又可对正常组织细胞造成毒副作用的结果。
[0099]
d acp-mmc ccdds的组织靶向性:acp-mmc ccdds腹腔注射后主要分布于腹膜间隙和淋巴组织。如图9所示,acp-mmc ccdds腹腔注射后,对种植于腹腔内的癌细胞在腹膜上形成一道严密的封锁线,使其不能通过腹膜在腹腔组织中形成转移灶;其大量地在淋巴组织中的分布使癌细胞不能通过淋巴道转移;即使通过血液转移的肿瘤,在进入血液之前,也必须通过组织液,而且只有在组织液中才能形成癌灶,因此组织液中存在的ccdds能有效地预防转移和复发。
[0100]
(4)实验结论:acp-mmc ccdds能对腹腔种植转移和复发进行有效地的预防和对抗,其机理是其组织靶向性和浓度选择性。
[0101]
表5:药物对人肝癌hepg-2 细胞裸鼠移植癌转移灶数的影响
表5中mean
±
sd,n = 6。腹腔注射给药。mmc:丝裂霉素;cbmc:acp-mmc ccdds的一种制剂。*p 《 0.05,*p 《 0.01,***p 《 0.001 与对照组相比;
△△
p ﹤ 0.01;
△△△
p ﹤ 0.001与mmc组相比。发生率的比较用检验,转移率的比较用t 检验。
[0102]
实施例七:acp-dox ccdds对人h460肺癌的实验性治疗:(1) 模型制备:采用荧光素酶pcibap-luc-9基因转染的h460人肺鳞癌细胞株(l-h460)对babl/c-nu裸小鼠进行胸腔种植。取处于对数生长期的l-h460细胞株,将其消化分散成单细胞悬液,4℃ 离心后以生理盐水调节其浓度为5
×
106细胞/ml,以0.2 ml/只胸腔接种c57bl/6小鼠,6 d后癌灶形成。
[0103]
(2) 动物分组及给药:制备的模型动物36 只,雌雄各半,18 ~ 22 g,平均分为8组:生理盐水对照1组,空白acp(5 mg/kg,为含有1 mg/kg dox的载体剂量)对照1组,dox 0.33、1和 3 mg/kg 3 个剂量组, acp-dox ccdds (以其所含的dox 剂量计)0.33、1 和 3 mg/kg 3 个剂量组。瘤细胞接种 24 h后称重,随机分组按上述剂量一次性胸腔注射。对照组给予0.2 ml/只生理盐水胸腔注射。空白acp 对照组给予未载药的acp 5 mg/kg 胸腔注射。
[0104]
(3)治疗效果检测:a对荷瘤小鼠生存时间的影响:给药后观察各组小鼠的死亡时间及死亡数,生存时间超过30天按照30天计算,待各组小鼠全部死亡后,按照公式延长率 = (用药组生存时间—对照组生存时间)/对照组生存时间
´
100% 计算生命延长率,统计处理。
[0105]
b对肿瘤生长的影响:用ivis成像系统观察肿瘤细胞的生长及癌灶形成情况。分别于给药后d5、d10和d15对肿瘤大小进行体内影像学观测,以产生的荧光强度对肿瘤大小进行定量,用spss13.0软件进行组间差异的方差分析。抑瘤率(%)=(空白对照组平均光量子强度-实验组平均光量子强度)/对照组平均光量子强度
×
100%;并通过动态图像观测肿瘤的转移情况。
[0106]
(4)实验结果:a生命延长率:在小鼠h460人肺癌模型上,载体acp 没有明显的延长生命作用。如表6所示,dox 和acp-dox ccdds均可显著延长h460肺癌小鼠的生存时间,具有明显的剂量-效应关系,经检验(p﹤0.001),具有统计学意义。acp-dox ccdds对生命的延长率明显大于
dox。阴性对照组小鼠平均生存天数为14.2天,在剂量为 0.33 mg/kg 时dox 对小鼠的生命有延长作用倾向,但没有统计学意义,acp-dox ccdds对小鼠的生命延长率与对照组相比具有统计学意义。在剂量为1 mg/kg 时,dox 的生命延长率为42.9%,acp-dox ccdds生命延长率为71.4%,两组的生命延长率之间有统计学显著性。在实验所用最大剂量 3 mg/kg 情况下,dox 的生命延长率为78.6%,但并不能使免于死亡,而acp-dox ccdds在相同剂量下能使所有的小鼠在30天观察时间内无死亡,生命延长率为110.5%(p﹤0.001),其生命延长作用显著高于dox。
[0107]
b对肿瘤的抑制作用如图 10所示:在用荧光素基因转染的人h460 肺癌细胞制备的肺癌模型的活体成像可见acp 组与对照组的肿瘤大小没有明显差异。dox 和acp-dox ccdds治疗组肿瘤都较对照组肿瘤明显为小,但acp-dox ccdds治疗组肿瘤较等剂量dox 治疗组肿瘤更小。如表7所示,利用光量子数进行定量分析表明,dox 和acp-dox ccdds对h460 细胞实验性肺癌都能明显抑制肿瘤生长,具有统计学意义,但acp-dox ccdds的作用较 dox 更强。acp-dox ccdds不同于 dox 的明显特征是在第15天,对肿瘤的抑制率仍在明显上升,而 dox 抑制率已不再进一步上上升:在 1 mg/kg 剂量下,于给药后第5 天,acp-dox ccdds的荧光强度为dox 的 1/2,第 10天,为 dox 的 1/3,第 4 天则只有 1/4,提示acp-dox ccdds对h460细胞癌的抑制作用远大于dox而且药效持久存在,反映acp-dox ccdds对 dox 的缓释作用,使之抗肿瘤作用强大而持久。
[0108]
(5)结论:acp-dox ccdds对人肺癌具有良好的治疗效果。可明显抑制肿瘤生长,延长荷瘤动物的生命。
[0109]
表6:药物对人h460肺癌小鼠生存时间的影响表7:药物对肿瘤荧光量子数的影响
表6中mean
±
sd,n = 6。胸腔注射给药。dox:紫杉醇;ncdox:acp-acp-dox ccdds ccdds的一种制剂。
* p﹤0.05,
** p﹤0.01, *** p﹤0.001与生理盐水组相比;
△
p﹤0.05;
△△△
p﹤0.001与dox 组相比。
[0110]
表7中mean
±
sd, n = 6。胸腔注射给药。control:对照;acp:活性炭;dox:紫杉醇;ncdox:acp-acp-dox ccdds ccdds的一种制剂。* p﹤0.05,**p﹤0.01,
*** p﹤0.001与对照组相比;
△
p﹤0.05;
△△
p﹤0.01与mmc 组相比。
[0111]
综上所述,通过本发明制备的组织释药浓度可控的抗癌药物递送系统的载药量qe和载体粒子的表面浓度ce是精确可控的。因为组织释药浓度可控的抗癌药物递送系统在组织中释放药物,产生的药物浓度是qe和ce的函数,因此确定的qe和ce必定产生确定的组织药物浓度。因此组织释药浓度可控的抗癌药物递送系统的可控性制备为组织释药浓度可控的抗癌药物递送系统在组织产生的药物浓度的精确可控奠定了基础。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1