一种基于单细胞转录组测序的物种间同源基因转换分析方法及系统与流程
本发明属于单细胞转录组测序,涉及一种基于单细胞转录组测序的物种间同源基因转换分析方法及系统。
背景技术:
1、单细胞转录组测序是在单个细胞水平进行高通量转录组测序的一项技术,可以研究单个细胞内的基因表达情况和细胞间的异质性。结合一些高级分析内容,例如,scenic转录因子调控分析、cellchat细胞通讯分析、scran细胞周期分析等,能够在单细胞水平上进一步揭示基因表达调控机制、细胞间相互作用关系以及发育过程中的潜在分子机制。
2、在单细胞转录组测序数据分析领域,许多高级分析均高度依赖于物种特定的数据库,而且可用的数据库多集中于人、小鼠或果蝇。例如,scenic转录因子调控分析的官方数据库仅支持人、小鼠和果蝇三个物种;cellchat细胞通讯分析的官方数据库仅支持人和小鼠两个物种;scran细胞周期分析的数据库同样仅支持人和小鼠。许多非常规物种受限于背景数据库,无法直接利用这些高级分析来进行数据挖掘。因此,需要采用物种间同源基因转换的方法——也就是将研究物种的基因,通过基因序列相似性比对的方式,转换为有数据库支持的物种基因来进行分析,这一方法对于特殊物种在单细胞测序领域的研究具有重要意义。
3、现有的单细胞转录组数据分析流程中,尚未建立物种间同源基因一键化转换分析方法和流程,只能依靠手动处理。手动处理的缺陷在于:(1)操作步骤多,分析过程繁琐,易出错;(2)自动化程度低,低效费时,无法满足快速对特殊物种进行高级分析的需求;(3)对分析人员的编程能力要求高,且不利于数据结果复现。因此,需要建立一个针对单细胞转录组数据进行同源基因一键化转换的分析方法流程。
技术实现思路
1、为了解决现有技术存在的不足,本发明的目的是提供一种基于单细胞转录组测序的物种间同源基因转换分析方法,能够快速对不同物种间的同源基因进行转换,解决单细胞转录组高级分析中的物种数据库限制问题,从而使特殊物种也能直接利用高级分析进行下游数据挖掘。
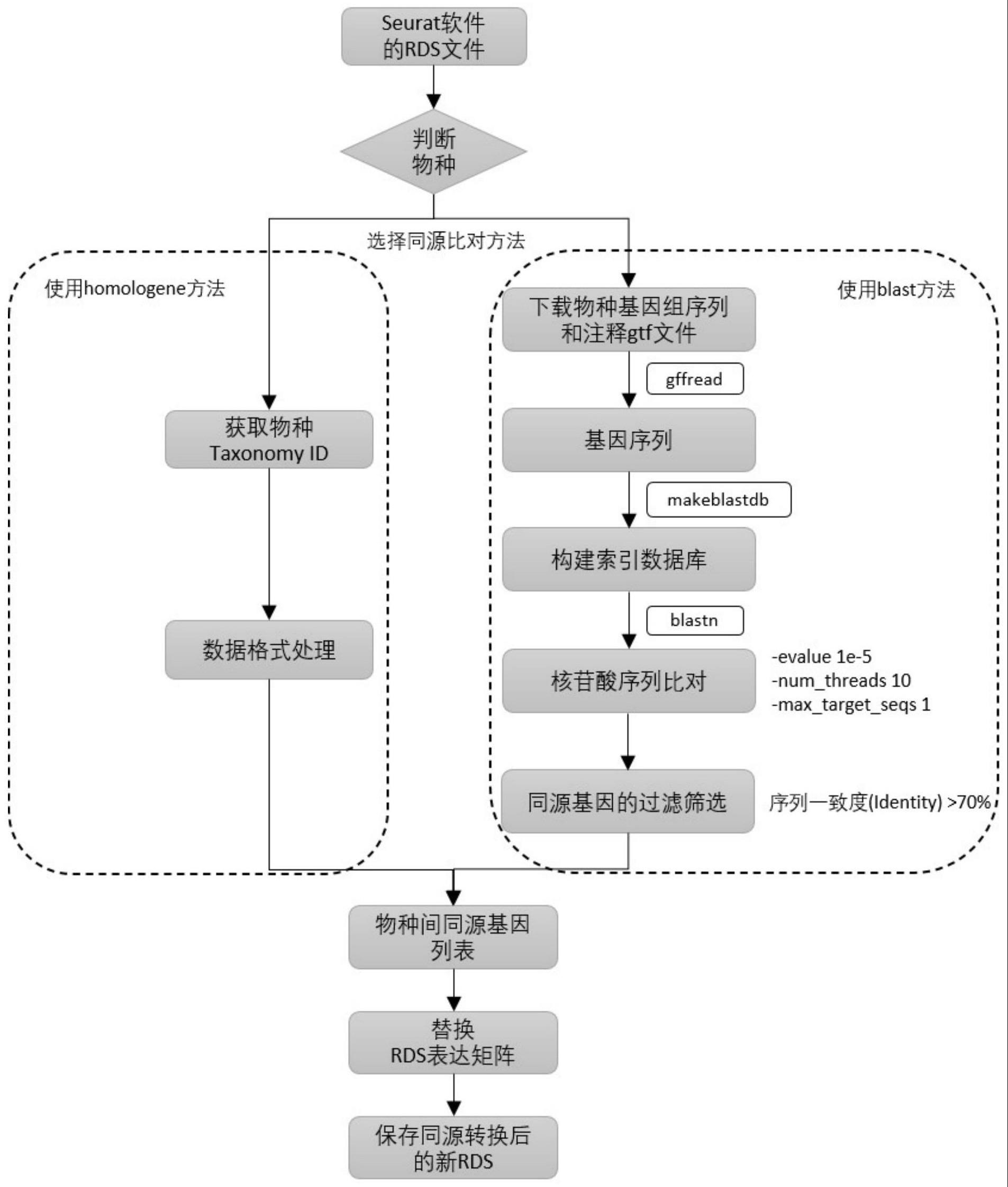
2、为了实现上述目的,本发明提出了一种基于单细胞转录组测序的同源物种转换分析方法,包括以下步骤:
3、步骤一、文件准备:
4、准备单细胞转录组标准分析软件seurat产生的待转换物种的rds文件。
5、步骤二、物种判断及同源比对方法选择:
6、根据需要做同源转换的物种拉丁名,判断其是否存在于以下21个模式生物列表中:人(homo sapiens)、小鼠(mus musculus)、大鼠(rattus norvegicus)、黑猩猩(pantroglodytes)、猕猴(macacamulatta)、犬(canis lupus familiaris)、牛(bos taurus)、鸡(gallus gallus)、斑马鱼(danio rerio)、黑腹果蝇(drosophila melanogaster)、热带爪蟾(xenopus tropicalis)、秀丽线虫(caenorhabditis elegans)、冈比亚按蚊(anophelesgambiae)、拟南芥(arabidopsis thaliana)、水稻(oryza sativa)、乳酸克鲁维酵母(kluyveromyces lactis)、棉假囊酵母(eremothecium gossypii)、粟酒裂殖酵母(schizosaccharomyces pombe)、酿酒酵母(saccharomyces cerevisiae)、稻瘟病菌(magnaporthe oryzae)、粗糙链孢霉(neurospora crassa)。若存在,则默认使用homologene方法进行同源基因转换;若不存在,则默认使用blast方法进行同源基因转换。
7、步骤三、使用homologene方法进行同源基因转换:
8、r语言的homologene软件包中收录有21个模式物种之间的44233组同源基因,当待转换的物种存在于步骤二中的这21个模式物种内,根据需要查找同源基因的基因列表(下文中用gene表示)、待转换的物种编号taxonomy id(下文中用intaxid表示)以及需要转换成为的物种编号taxonomy id(下文中用outtaxid表示),使用homologene(gene,intax=intaxid,outtax=outtaxid)函数命令,具体操作为:利用ncbi网站中的homologene数据库,该数据库收集了21个物种的同源基因数据,可以通过基因名称来查询物种间对应的同源基因,获得物种间同源基因列表。
9、步骤四、使用blast方法进行同源基因转换:
10、当待转换的物种不存在于步骤二的21个模式物种列表内,使用blast方法进行同源基因转换。下载待转换物种和目标物种的基因组序列和注释gtf文件后,利用blast软件进行序列相似性比对,获得待转换物种与目标物种基因组序列的相似程度,筛选保留期望阈值evalue≤1e-5,且序列一致度(identity)>70%的同源基因对,当每条待转换物种的基因序列筛选得到多条目标物种的同源基因时,选取序列一致度(identity)最高的同源基因对,作为最优的一条比对结果,获得物种间同源基因列表。
11、其中,期望阈值的设置可以用来说明比对结果的可靠性:即在随机情况下,其他序列与目标序列相似度大于该结果序列的可能性,因此其分值越低越好。序列一致度可以用于说明待转换物种与目标物种基因组序列之间的一致程度。
12、所述序列相似性比对是指利用两条序列之间的核酸碱基差异来测定序列之间的相似性。两条序列中相应位置的核酸碱基如果差异大,那么序列的相似性低,反之,序列的相似性就高。通过检测序列之间核酸碱基的相似度,从而判断序列间的同源性。
13、步骤五、替换rds表达矩阵:
14、根据物种间同源基因列表,在步骤一待转换物种的rds文件中剔除未比对上的基因名,利用比对上的同源基因对待转换物种的rds文件中的表达矩阵进行替换,包括原始counts表达矩阵和标准化后的表达矩阵。
15、所述未比对上的基因是指经过序列相似性比对后,基因的期望阈值evalue>1e-5,和/或,序列一致度(identity)≤70%;
16、所述表达矩阵是指行为基因,列为细胞的表达量表格。依据获得的物种间同源基因列表,将待转换物种表达矩阵的基因名(即行名)替换为对应目标物种的同源基因名。
17、所述原始counts表达矩阵是指单细胞转录组测序中的表达量原始计数矩阵,标准化后的表达矩阵是指基于表达量原始计数矩阵进行log化处理,目的是消除测序深度和/或文库大小的影响。原始counts表达矩阵和标准化后的表达矩阵的格式相同,均为行是基因,列是细胞。
18、步骤六、保存同源转换后的新rds:
19、使用saverds函数将替换完同源基因的文件保存为新rds,该文件可无缝衔接单细胞转录组的下游高级分析,直接应用于scenic转录因子调控、cellchat细胞通讯和scran细胞周期等依赖特定物种数据库的分析内容。
20、优选地,所述步骤三和步骤四为可选的并行步骤:
21、当需要转换的物种已被收录在homologene软件包中时(homologene软件包中包含步骤二中21个模式物种的同源基因),本发明默认选择homologene方法进行同源基因转换,根据已收录的同源基因关系对直接进行快速转换,最大程度上缩短运行时间。当需要转换的物种未被收录在homologene软件包中时(即需要转换的物种不包含在步骤二中的21个物种中),默认选择blast方法进行同源基因转换,对物种类型没有限制,只需要下载物种基因组序列和注释gtf文件,即可进行转换。
22、优选地,所述步骤三中包括如下步骤:
23、使用r语言homologene软件包中的taxdata函数,自动获取待转换的以及需要转换成的物种的对应编号(taxonomy id),根据待同源转换的基因列表和物种编号,使用homologene函数得到同源基因对表格,去除重复的基因名称,获得物种间同源基因列表。
24、优选地,所述步骤四中包括如下步骤:
25、根据下载的待转换物种基因组序列和注释gtf文件,利用gffread软件分别提取出待同源转换物种和目标物种的基因序列文件。通过makeblastdb软件对目标物种的基因序列构建核苷酸索引数据库后,利用blastn软件对核酸序列进行相似性比对,筛选期望阈值evalue≤1e-5,且序列一致度(identity)>70%的同源基因对,使用10个线程并行运行(num_threads 10),指定输出格式(outfmt)为6(即输出含有12列内容的表格,表头分别为:qseqid查询序列的标识、sseqid比对上的目标序列的标识、pident一致性百分比、length比对区域的长度、mismatch比对区域的错配数、gapopen比对区域的空缺数目、qstart比对区域在查询序列上的起始位点、qend比对区域在查询序列上的终止位点、sstart比对区域在目标序列上的起始位点、send比对区域在目标序列上的终止位点、evalue比对结果的期望值、bitscore比对结果的打分),针对每个查询序列选取最优的一条比对结果(max_target_seqs 1),获得物种间同源基因列表。
26、本发明还提供了上述方法在其他高通量组学数据分析中的应用。本发明同时支持在没有rds文件的情况下(常见于非单细胞转录组测序领域),仅提供物种信息和待转换基因即可获得同源基因列表,具体方法与步骤三和步骤四一致,获得的同源基因列表可以用于其他高通量组学数据,如普通转录组测序领域的同源转换分析,不局限于单细胞转录组测序。
27、本发明还提出了一种实现上述物种间同源基因转换分析方法的转换分析系统,所述系统包括:最优方法选择模块、homologene同源转换模块、blast同源转换模块、以及rds同源基因替换模块。
28、所述最优方法选择模块是指根据物种特征,选择homologene或blast最优方法进行同源转换;
29、所述homologene同源转换模块是指利用homologene软件包,自动获取待转换物种和目标的物种编号(taxonomy id)后,通过homologene函数得到同源基因对;
30、所述blast同源转换模块是指利用gffread软件提取基因序列后,通过blast软件进行序列比对,根据预设阈值筛选同源基因对;
31、所述rds同源基因替换模块是指根据同源基因对,将待转换物种rds中的表达矩阵基因名替换为目标物种的同源基因名,获得新rds文件。
32、本发明的有益效果包括:
33、1.填补了单细胞转录组分析领域一键化进行物种间同源基因转换分析方法流程的空缺:目前基于单细胞转录组测序做物种间同源基因转换,没有一套系统性的流程,只能依赖繁琐的手动处理。例如当物种同源基因未被整理收录时,使用blast方法需要手动整理出基因序列文件,使用特定条件逐一筛选出同源基因列表,还要手动读入rds文件替换表达矩阵等,整个过程十分繁琐,操作步骤多,非常消耗人工,同时对操作人员的编程能力有一定要求。本发明优化了操作步骤,只需要提供rds文件、物种基因组序列和注释gtf文件(物种基因组序列和注释gtf文件仅在选用blast方法时需提供),流程会根据是否提供了基因组序列和注释gtf文件,自动选择blast方法调用gffread软件整理提取基因序列,并通过预先设置的阈值筛选同源基因对,获得同源基因转换后的新rds文件。本发明首次提出基于单细胞转录组测序的同源基因转换分析方法和系统,适用于所有有参考基因组的物种,无其他特殊限制,兼容性广,有利于结果复现,并且适配度高,同源转换后的rds文件可以和单细胞转录组下游高级分析无缝衔接,实现一键化高效分析。
34、2.结合了两种同源基因转换分析方法、同时优化了blast的数据处理过程,提升分析效率:本发明将homologene和blast两种方法结合起来进行同源基因转换分析,homologene方法可以根据已收录物种的同源基因对,快速获得同源基因列表,运行速度相比blast方法快至少2-3倍,但支持转换的物种类型有限;而blast方法没有物种类型限制,只需要有物种基因组序列和注释gtf文件,即可进行转换分析。另外,本发明还优化了blast分析的数据处理过程,无需人工整理注释gtf文件、基因序列文件、构建索引库和筛选最优结果,只要提供基因组序列和注释gtf文件,就能准确获得同源基因列表。相比现有方法,分析时间可以缩短至少1-2小时,分析效率能够提升至少2倍。
35、3.拓宽了单细胞转录组高级分析的应用范围:单细胞转录组测序的高级分析由于物种数据库限制,大多只能应用于模式物种。使用本发明的方法流程可以使得特殊物种直接应用高级分析进行下游数据挖掘,克服数据库受限问题,对于特殊物种在单细胞测序领域的研究具有重要意义。
- 还没有人留言评论。精彩留言会获得点赞!