一种识别空间转录组空间区域和细胞类型的特征学习方法
本发明涉及生物信息学领域,更具体地,涉及一种识别空间转录组空间区域和细胞类型的特征学习方法。
背景技术:
1、空间转录组测序是一种新兴的测序技术,它能够以多细胞、单细胞、甚至亚细胞分辨率提供带有空间坐标位置的转录组信息,为细胞功能、表型和组织微环境中位置的关系提供了重要信息,而解析这些问题的第一步就是正确划分空间区域。
2、划分空间区域的关键在于如何学习单个细胞的特征,以及如何融合空间位置信息。现有的多个方法可以进行空间区域的划分。其中bayesspace(zhao,e.et al.“spatialtranscriptomics at subspot resolution with bayesspace.(使用bayesspace的亚斑点分辨率空间转录组学)”nat biotechnol 39,1375-1384,2021)使用带有马尔可夫随机场的贝叶斯模型来实现空间聚类;spagcn(hu,j.et al.“spagcn:integrating geneexpression,spatial location and histology to identify spatial domains andspatially variable genes by graph convolutional network.(spagcn:整合基因表达、空间位置和组织学,通过图卷积网络识别空间域和空间可变基因)”nat methods 18,1342-1351,2021)使用图卷积网络整合基因表达,空间位置以及组织图像来识别空间域;stagate(dong,k.&zhang,s,“deciphering spatial domains from spatially resolvedtranscriptomics with an adaptive graph attention auto-encoder.(使用自适应图注意力自动编码器从空间分辨的转录组学中破译空间域)”nat commun 13,1739,2022)使用自适应的图注意力自编码器学习空间区域。
3、现有方法的主要问题是:(1)对空间位置的利用不够,导致区域划分效果有限。现有方法仅基于每个细胞与相邻细胞的表达相似性聚合邻居信息,这是对表达数据的过度利用,从而造成表达相似细胞的过度平滑。(2)没有数据的测序噪声进行显式的建模,这使得这些方法难以处理稀疏程度高的空间转录组数据。但随着测序技术的发展,数据的分辨率逐渐升高,稀疏程度也随之提高,建模数据的测序噪声也变得尤为重要。(3)对于兼具单细胞分辨率的空间转录组数据,不能同时识别空间区域和细胞类型,使得数据分析流程复杂化。
技术实现思路
1、本发明提供一种识别空间转录组空间区域和细胞类型的特征学习方法,同时识别空间转录组数据的空间区域和细胞类型,还保留了明确的生物学信号,可以实现良好的低维特征可视化和正确的轨迹推断。
2、为解决上述技术问题,本发明的技术方案如下:
3、一种识别空间转录组空间区域和细胞类型的特征学习方法,包括以下步骤:
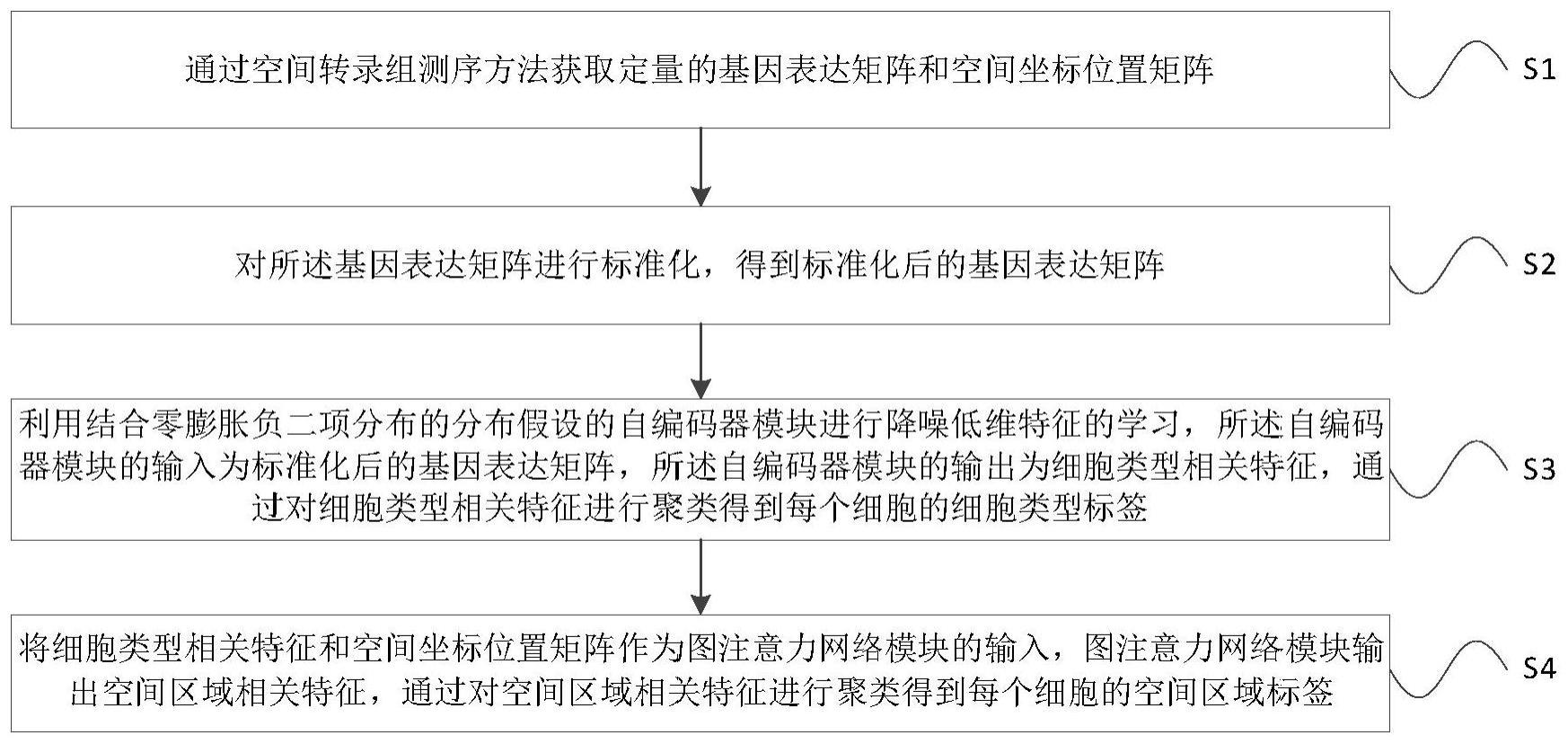
4、s1:通过空间转录组测序方法获取定量的基因表达矩阵和空间坐标位置矩阵;
5、s2:对所述基因表达矩阵进行标准化,得到标准化后的基因表达矩阵;
6、s3:利用结合零膨胀负二项分布(zero-inflated negative binomialdistribution,简称zinb)的分布假设的自编码器(autoencoder,简称ae)模块进行降噪低维特征的学习,所述自编码器模块的输入为标准化后的基因表达矩阵,所述自编码器模块的输出为细胞类型相关特征(cell type-related embedding,简称ce),通过对细胞类型相关特征进行聚类得到每个细胞的细胞类型标签;
7、s4:将细胞类型相关特征和空间坐标位置矩阵作为图注意力网络(graphattention network,简称gat)模块的输入,图注意力网络模块输出空间区域相关特征(spatial domain-related embedding,简称se),通过对空间区域相关特征进行聚类得到每个细胞的空间区域标签。
8、优选地,步骤s1中基因表达矩阵和空间坐标位置矩阵,具体为:
9、基因表达矩阵x中的每行代表一个测序基本单元,包含单个细胞或多个细胞,一共有n个细胞,基因表达矩阵x中的每列代表一个基因,一共有g个基因;
10、空间坐标位置矩阵y的每行代表一个细胞,空间坐标位置矩阵y的列是该细胞的二维或三维位置信息。
11、优选地,步骤s2中对所述基因表达矩阵进行标准化,具体为:
12、
13、式中,xn表示细胞n的基因表达向量,为所述基因表达矩阵的第n行,sn表示细胞n的文库大小,sf表示标准化后所有细胞的文库大小(library size),表示标准化后的细胞n的基因表达向量。
14、优选地,所述基因表达矩阵中的每个元素都遵循零膨胀负二项分布,其参数为(πng,rng,pg),其中πng是观察到真实基因表达值为0的概率,(rng,pg)是负二项分布的标准参数,xng是在达到rng次失败时的成功次数,pg是每个伯努利试验的失败概率。
15、优选地,xng的似然函数为:
16、
17、其中,δ0()表示狄拉克函数,γ()表示伽马函数。
18、优选地,步骤s3中自编码器模块包括两层编码器和两层解码器,表示为:
19、
20、z′=f2(z)
21、r′=f3(z′)
22、π=f4(z′)
23、式中,f1是编码器,编码器包含两层,分别将输入数据从g维降维至m′维、从m′维降维至m维,表示标准化后的基因表达矩阵,z为降维后的细胞类型相关特征矩阵;f2、f3、f4分别为解码器的三个输出,解码器包括两层,分别将输入数据从m维升维至m′维、m′维升维至g维,z′为解码器的第一个输出,f3、f4分别作用于z′以学习参数r′和参数π,参数r和参数π分别为n×g维的矩阵,分别由元素rng和πng组成,r′由元素r′ng组成,rng=s′nr′ng,其中s′n是细胞n的缩放系数,s′n=exp(log10sn)。
24、优选地,由元素lg组成的参数向量l是通过指数变换从可学习向量获得的,具体为:
25、
26、式中,lg是负二项分布的logit。
27、优选地,所述自编码器模块的损失函数定义为负二项分布的负对数似然函数(negative log-likelihood function,简称nll):
28、loss1=nllzinb(x;π,r,p)
29、式中,nllzinb()表示负二项分布的负对数似然函数。
30、优选地,步骤s4具体为:
31、首先根据各个细胞的位置构建对称的邻接矩阵a,节点的集合写成v={v1,v2,...,vn},那么邻接矩阵a的每个元素aij可以表示为:
32、
33、式中,是节点vj的近邻节点集合,通过knn或距离截断来计算;
34、构建相似性矩阵∑,令细胞之间的空间区域相关特征相关性随着距离的增加而减少,并趋于指数衰减,所以理想情况下∑是基于高斯核的n×n维空间相关矩阵:
35、
36、式中,γ表示带宽参数;
37、所述图注意力网络模块包括两个图注意层,令图注意层的输入是n×n维的特征矩阵为h=(h1,h2,...,hn),输出为n×n′维的矩阵h′=(h′1,h′2,...,h′n),则:
38、
39、式中,w是n′×n维的权重矩阵,是vj邻点的集合,αij是使用softmax函数的归一化注意力系数矩阵:
40、
41、eij=at(whi||whj)
42、其中a是可学习向量,||是连接操作,在图注意层中使用指数线性单元作为激活函数σ;
43、空间区域相关特征u经过学习,可以表示为:
44、u=gat2(gat1(z))
45、式中,gat1()、gat2()分别表示第一层图注意层和第二层图注意层。
46、优选地,所述图注意力网络模块的目标函数为:
47、loss2=λspatial*lspatial+λrec*lrec
48、式中,lspatial=mse(uut,∑),lrec=mse(u,z),λspatial和λrec为权重参数。
49、与现有技术相比,本发明技术方案的有益效果是:
50、(1)本发明融合了统计分布假设和深度学习算法,使得模型兼具统计建模的表征能力和深度学习的学习效率,显著提高了空间转录组数据的区域划分能力和特征学习效果。
51、(2)本发明的ae模块的建模学习到了精细可靠的细胞类型相关特征,因此对于具有单细胞分辨率的空间转录组数据,可以同时输出细胞类型和空间区域的标签,简化了空间转录组数据的分析步骤。
52、(3)本发明的gat模块充分利用空间位置信息,利用全局的位置信息来防止局部过拟合的出现。同时,可以自适应地更新低维表征,保留了数据的生物学意义,学习到可解读的特征空间和可靠的轨迹推断结果。
- 还没有人留言评论。精彩留言会获得点赞!