基于深度学习与原子间作用力加权的分子对接评分方法与流程
本发明属于计算机辅助药物分子设计,具体涉及一种基于深度学习与原子间作用力加权的分子对接评分方法。
背景技术:
1、分子对接是计算机辅助药物设计的一种方法,旨在预测药物分子与其作用靶点之间的最佳结合方式,为药物研发提供指导。分子对接基于计算机模拟配体和受体分子的结构,以寻找它们之间的最佳空间排列方式,并评估分子的结合能力。精确的分子对接预测将大大降低药物研发成本,加速药物研发进程。
2、传统的分子对接方法具有评分函数简单、计算速度快的优点,但其对于复杂构象的评分精度有限,难以精确预测分子的结合能力。在非经验情况下,其精度更是有限。因此,传统方法往往难以满足大规模虚拟筛选的需求。
3、随着蛋白质三维结构数据的快速增加,基于深度学习的分子对接方法成为热门研究领域。基于深度学习的方法可以自动从数据中提取特征,具有强大的表征能力。通过对蛋白质-配体复合物的3d空间结构进行建模,神经网络可以提取有效的小分子和蛋白质的表征。然而,分子对接需要使用大量的配体在蛋白质空间内平移旋转的中间构象数据,目前这样的数据集仍然非常稀少。因此,如何利用现有的蛋白质-配体晶体数据,扩充基于深度学习的分子对接方法所需要的数据,并训练出一个更准确、泛化能力更好的分子对接评分方法,仍然是一个具有挑战性的问题。
技术实现思路
1、为解决公知技术中存在的以上不足,本发明旨在提供一种基于深度学习与原子间作用力加权的分子对接评分方法,通过利用已知的蛋白质-配体复合物晶体数据,采用分子构象搜索和采样算法扩充训练数据集,以训练出一个更准确,泛化能力更好的分子对接评分方法。
2、为实现上述目的,本发明所采用的技术方案如下:
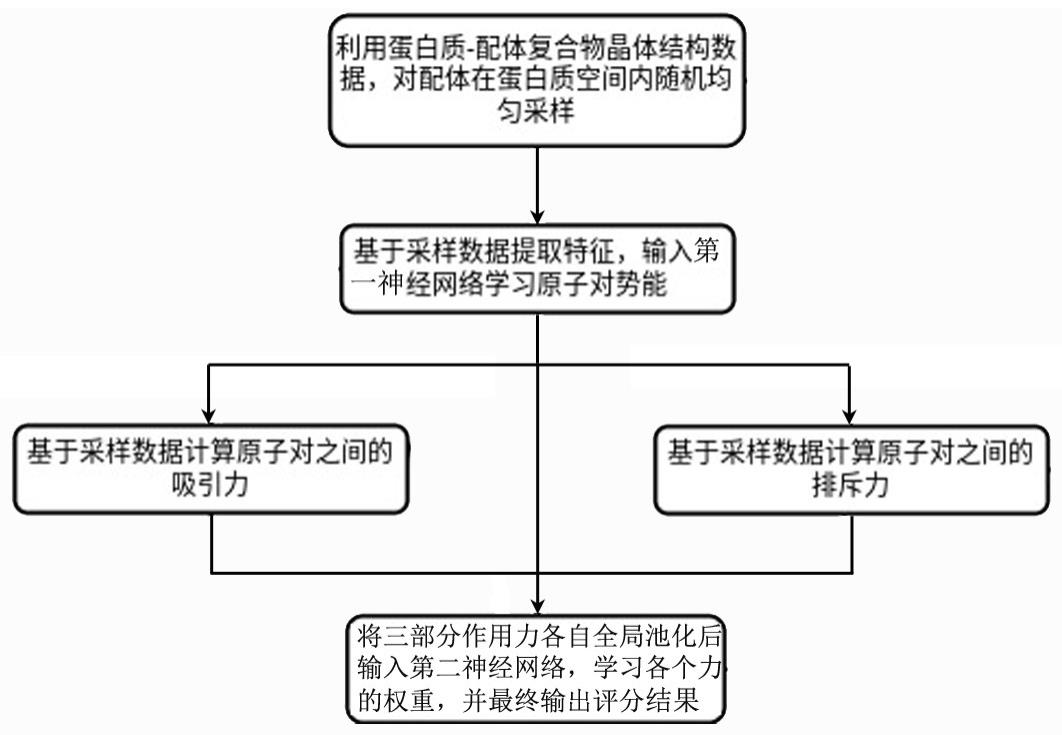
3、一种基于深度学习与原子间作用力加权的分子对接评分方法,该方法包括依次进行的以下步骤:
4、s1、采用构象采样搜索算法对蛋白质-配体晶体数据随机均匀采样得到采样后的数据集;
5、s2、对采样后的数据集重新定义标签得到可训练的数据集;
6、s3、对可训练的数据集进行第一特征提取,将提取的第一特征形成原子对特征表达后,输入第一神经网络进行学习,得到配体与蛋白质的原子对之间的势能预测值;
7、在此过程中,获得邻接矩阵;
8、s4、对可训练的数据集进行第二特征提取,并通过步骤s3中得到的邻接矩阵获取数据,将数据分别输入吸引力函数和排斥力函数,分别得到配体与蛋白质的原子对之间的吸引力预测值和排斥力预测值;
9、s5、根据配体与蛋白质的原子对之间的势能预测值、吸引力预测值和排斥力预测值,分别通过全局池化获得蛋白质-配体的势能预测值、吸引力预测值和排斥力预测值;
10、s6、将蛋白质-配体的势能预测值、蛋白质配体的吸引力预测值和蛋白质-配体的排斥力预测值输入第二神经网络,学习各项权重,计算损失函数后,利用反向传播算法更新模型权重;
11、s7、重复步骤s3~s6进行迭代,直至满足迭代停止条件,训练过程完成,利用训练完成的模型预测蛋白质-配体的对接构象评分。
12、作为限定,所述步骤s1中采用构象采样搜索算法对蛋白质-配体晶体数据随机均匀采样过程通过以下步骤依次进行:
13、a1)以蛋白质-配体复合物结晶位置为原点,配体在蛋白质空间内随机平移旋转;
14、a2)计算配体平移旋转后的位置构象与原结晶位置构象之间的rmsd,所述rmsd为均方根偏差;
15、a3)将得到的rmsd作为加权系数,对蛋白质-配体结晶位置的标签值进行加权处理,得到配体在平移旋转后的构象的标签值;
16、a4)重复上述步骤a1)~步骤a3),得到多个rmsd以及与rmsd相对应的标签值;
17、a5)将配体构象按照rmsd的大小划分为不同的区间,每个区间保存相同个数的配体构象,所保存的配体构象组成采样后的数据集。
18、作为第二种限定,根据以下公式重新定义标签的方法为:
19、
20、其中,kd表示解离常数,反映的是化合物对靶标的亲和力大小,值越小亲和力越强;ki表示抑制常数,反映的是抑制剂对靶标的抑制强度,抑制常数越小说明抑制能力越强;-logkd/ki是指对kd或者ki取负对数;
21、原结晶位置构象以及的配体构象的标签为蛋白质-配体结晶位置的标签值;的配体构象的标签为晶体构象的标签值以及所对应的rmsd的比值。
22、作为第三种限定,所述第一特征包括第一节点特征和第一边特征;所述第一节点特征包括配体分子的原子特征以及距离配体原子内的蛋白质原子的特征;所述第一节点特征的类型包括:原子类别、原子的邻域原子类型、原子的坐标、和原子的范德华半径;
23、所述第一边特征包括配体分子内有共价键连接的共价键的键长,以及距离配体原子范围内的蛋白质原子与配体原子所组成的虚拟边的边长;
24、配体的共价键和距离配体原子内范围内的蛋白质原子与配体原子所组成的虚拟边构成边的连接信息,边的连接信息组成邻接矩阵。
25、作为第四种限定,所述原子类别的定义包含以下几类:c、n、o、卤素、p、s、金属、疏水c、氢键供体n、氢键受体n、其他类型的n、既可能是氢键供体又可能是氢键受体的n、是氢键供体并且是其他类型的n、是氢键受体并且是其他类型的n、既是氢键供体又是氢键受体并且是其他类型的n、氢键供体o、氢键受体o、其他类型的o、既可能是氢键供体又可能是氢键受体的o、既是氢键供体又是其他类型的o、既是氢键受体又是其他类型的o、既可能是氢键供体又可能是氢键受体又是其他类型的o、氢键受体s、其他类型的s、既是氢键受体又是其他类型的s;
26、原子的邻域原子类型包含以下几种:c、o、n、cc、co、cn、nn、ccc、ccn、cno、cco、coo、ccf、cnn、nnn;其中同族元素硫认为是氧、磷认为是氮,卤素都认为是氟。
27、作为第五种限定,所述步骤s3中将提取的第一特征形成原子对特征表达的方法为:将第一节点特征根据邻接矩阵融合成初始原子对特征表达,将初始原子对特征表达与第一边特征融合,最后形成既有原子特征信息又有边信息的原子对特征表达;
28、所述融合是将两种信息向量合并为一个向量,融合的方法为相加、相减、拼接、卷积中的一种或两种以上组合。
29、作为第六种限定,所述步骤s4中的第二特征包括第二节点特征和第二边特征;所述第二节点特征包括配体分子的原子特征以及距离配体原子内的蛋白质原子的特征,所述第二节点特征为原子的范德华半径;
30、所述第二边特征为配体分子内有共价键连接的共价键的键长,以及距离配体原子范围内的蛋白质原子与配体原子所组成的虚拟边的边长。
31、作为第七种限定,所述步骤s4中的吸引力函数为:
32、
33、排斥力函数为:
34、其中,d为成对原子之间的边长,r1为原子对中第一原子的范德华半径,r2为原子对中第二原子的范德华半径。
35、作为第八种限定,所述步骤s2中输入第一神经网络进行学习和步骤s6中输入第二神经网络进行学习的学习步骤均包括多层非线性变换和线性变换。
36、作为第九种限定,所述步骤s7中的迭代停止条件包含:b1、达到预设置的训练次数;b2、损失降到阈值以内;b3、当训练集中的损失函数连续迭代一定次数不再减小。
37、由于采用了上述的技术方案,本发明与现有技术相比,所取得的有益效果是:
38、(1)本发明利用蛋白质-配体复合物晶体数据,通过构象采样搜索算法,随机均匀的采样出一批适合于分子对接的蛋白质-配体构象,并为其重新定义标签,极大的扩充了训练分子对接所需要的数据,此数据集更接近真实对接过程;
39、(2)本发明中将新数据分别送入蛋白质-配体势能预测部分、蛋白质-配体吸引力预测部分、蛋白质-配体排斥力部分,预测三种作用模式,并对三种作用模式做加权学习,有助于提高分子对接过程中蛋白质和配体之间的结合构象的评分能力;
40、(3)本发明使用原子的邻域原子类型信息,很好的解决了在对接过程中,配体分子构象的平移旋转导致第三方软件在读取当前配体构象时,某些原子属性不一致从而导致数据分布不一致的问题,例如某原子在对接过程中的杂化状态跟初始杂化状态不一致;采用原子的邻域原子类型,提供给算法模型学习的是当前原子的周边原子信息,对于配体分子来说,这些信息是不变的,从而保证了数据分布的一致性;
41、(4)本发明利用原子间吸引力预测和排斥力预测,来对模型预测的蛋白质-配体结合构象的质量评价做修正,提高了模型的泛化能力。
42、综上所述,本发明利用现有的蛋白质-配体晶体数据,能够扩充基于深度学习的分子对接方法所需要的数据,并可以训练出一个更准确、泛化能力更好的分子对接评分方法。
- 还没有人留言评论。精彩留言会获得点赞!