一种单细胞分析方法、装置、电子设备和存储介质与流程
本技术涉及生物,更具体地说,涉及一种单细胞分析方法、装置、电子设备和存储介质。
背景技术:
1、大部分dna细胞以染色体的形式紧密盘绕在细胞核内,在进行复制和转录时,高度折叠的染色体结构需要暴露出dna序列,这段暴露的区域叫做染色质开发区域,也叫开放染色质open chromatin,这个区域可以供转录因子和其他调控元件结合。暴露的染色质开发区域包含启动子、增强子、绝缘子和沉默子等顺式调控元件,其可以被反式作用因子接近结合的特性叫做染色质的可及性。染色质开放区域与基因表达类似,在不同的组织细胞中和细胞的不同时期会动态变化,呈现出时空变化特异性,因此,对于染色质开发区域的精确分析可为染色质可及性的研究提供数据基础。
技术实现思路
1、有鉴于此,本技术提供一种单细胞分析方法、装置、电子设备和存储介质,用于通过关联和注释方法对单细胞的染色质开发区域进行精确分析,以便为染色质可及性的研究提供数据基础。
2、为了实现上述目的,现提出的方案如下:
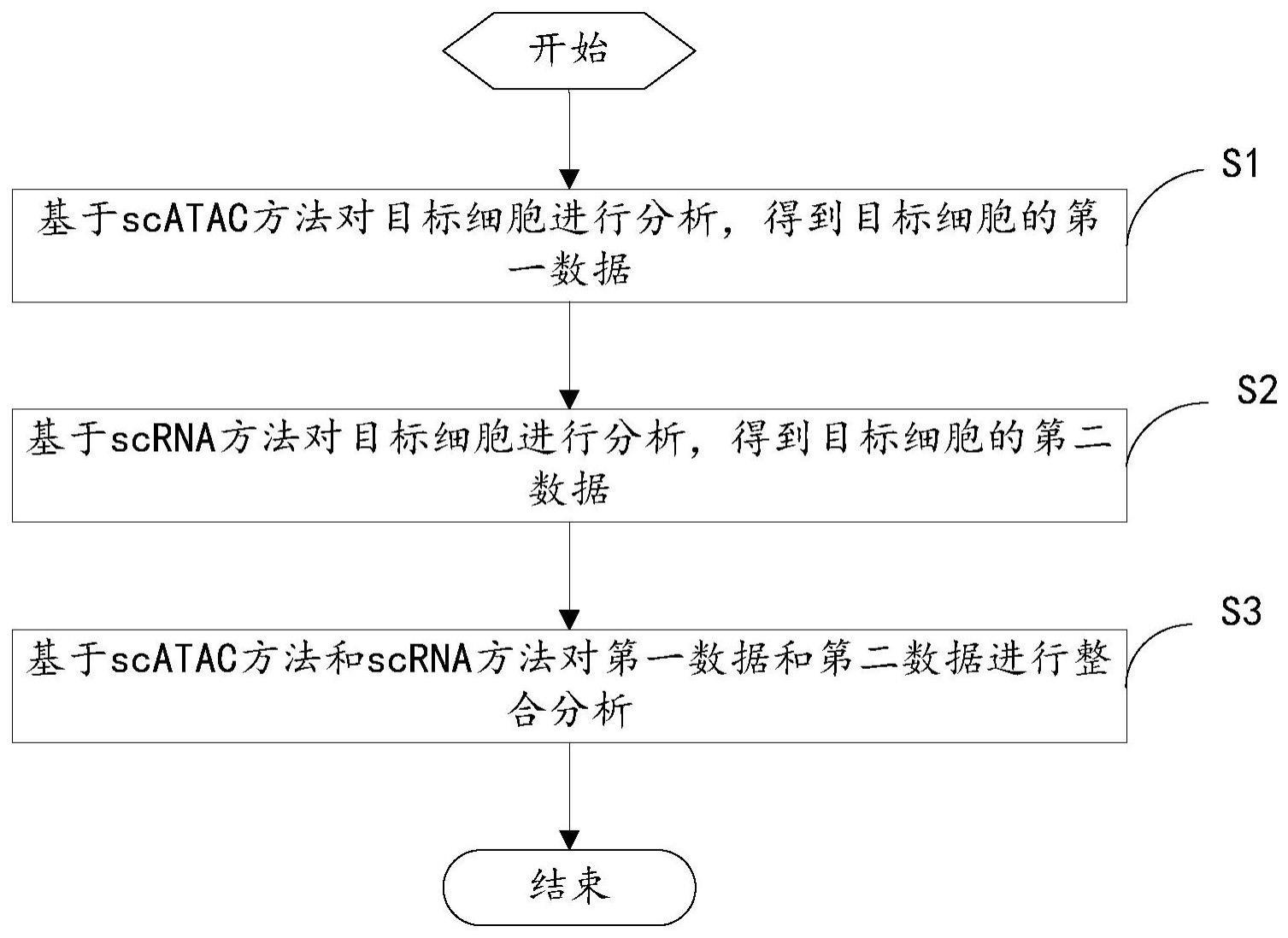
3、一种单细胞分析方法,应用于电子设备,所述单细胞分析方法包括步骤:
4、基于scatac方法对目标细胞进行分析,得到所述目标细胞的第一数据;
5、基于scrna方法对所述目标细胞进行分析,得到所述目标细胞的第二数据;
6、基于所述scatac方法和所述scrna方法对所述第一数据和所述第二数据进行整合分析,得到所述目标细胞的细胞类型和可及性。
7、可选的,所述基于scatac方法对目标细胞进行分析,得到所述目标细胞的第一数据,包括步骤:
8、基于所述scatac方法对所述目标细胞进行分析,得到所述目标细胞的启动子、增强子、沉默子和绝缘子的注释信息;
9、对所述注释信息进行质控处理;
10、对所述注释信息进行多样本数据合并;
11、对所述目标细胞的基因活性进行得分计算;
12、注释所述目标细胞的细胞类型;
13、对所述目标细胞的可及性进行分析。
14、可选的,所述对所述注释信息进行多样本数据合并,包括步骤:
15、采用archr方法读取每个样本的比对结果,将基因组上染色体区间按照预设字节分成连续没有交集的多个单元,统计细胞中每个单元的区间的数目,生成与所述多个单元相关的可及性矩阵;
16、或者,直接读取每个样本的比对结果,直接合并所述比对结果生成一个共同的峰值,然后重新计算每个样本在共同的峰值的可及性矩阵。
17、可选的,所述基于scrna方法对所述目标细胞进行分析,得到所述目标细胞的第二数据,包括步骤:
18、基于所述scrna方法对所述目标细胞进行合并分析;
19、基于所述scrna方法对所述目标细胞进行注释分析;
20、对所述目标细胞进行高级分析,得到所述第二数据。
21、可选的,所述基于所述scatac方法和所述scrna方法对所述第一数据和所述第二数据进行整合分析,得到所述目标细胞的细胞类型和可及性,包括步骤:
22、基于seurat cca方法将所述第一数据和所述第二数据映射到标准化的l2cca空间;
23、通过对拟时间邻近的多个所述目标细胞合并邻近多个细胞的策略,生成pseudobulk水平的基因表达、genescore、peaks和tf-motif可及性数据;
24、采用pearson相关性筛选显著的genescore-gene共表达的基因;
25、进一步筛选显著共表达genescore-gene关系对中基因的显著共表达peak-gene的关系对;
26、采用pearson相关性筛选tf-tfmotif可及性显著地共表达的转录因子;
27、结合tf-peaks的结合关系,筛选tf-target基因。
28、一种单细胞分析装置,应用于电子设备,所述单细胞分析装置包括:
29、第一分析模块,被配置为基于scatac方法对目标细胞进行分析,得到所述目标细胞的第一数据;
30、第二分析模块,被配置为基于scrna方法对所述目标细胞进行分析,得到所述目标细胞的第二数据;
31、整合输出模块,被配置基于所述scatac方法和所述scrna方法对所述第一数据和所述第二数据进行整合分析,得到所述目标细胞的细胞类型和可及性。
32、可选的,所述第一分析模块包括:
33、第一分析单元,被配置为基于所述scatac方法对所述目标细胞进行分析,得到所述目标细胞的启动子、增强子、沉默子和绝缘子的注释信息;
34、质控处理单元,被配置为对所述注释信息进行质控处理;
35、合并处理单元,被配置为对所述注释信息进行多样本数据合并;
36、活性计算单元,被配置为对所述目标细胞的基因活性进行得分计算;
37、类型注释单元,被配置为注释所述目标细胞的细胞类型;
38、第二分析单元,被配置为对所述目标细胞的可及性进行分析。
39、可选的,所述第二分析模块包括:
40、第三分析单元,被配置为基于所述scrna方法对所述目标细胞进行合并分析;
41、第四分析单元,被配置为基于所述scrna方法对所述目标细胞进行注释分析;
42、第五分析单元,被配置为对所述目标细胞进行高级分析,得到所述第二数据。
43、可选的,所述整合输出模块包括:
44、映射处理单元,被配置为基于seurat cca方法将所述第一数据和所述第二数据映射到标准化的l2cca空间;
45、数据生成单元,被配置为通过对拟时间邻近的多个所述目标细胞合并邻近多个细胞的策略,生成pseudobulk水平的基因表达、genescore、peaks和tf-motif可及性数据;
46、第一筛选单元,被配置为采用pearson相关性筛选显著的genescore-gene共表达的基因;
47、第二筛选单元,被配置为进一步筛选显著共表达genescore-gene关系对中基因的显著共表达peak-gene的关系对;
48、第四筛选单元,被配置为采用pearson相关性筛选tf-tfmotif可及性显著地共表达的转录因子;
49、第四筛选单元,被配置为结合tf-peaks的结合关系,筛选tf-target基因。
50、一种电子设备,包括至少一个处理器和与所述处理器连接的存储器,其中:
51、所述存储器用于存储计算机程序或指令;
52、所述处理器用于执行所述计算机程序或指令,以使所述电子设备实现如上所述的单细胞分析方法。
53、一种存储介质,应用于电子设备,所述存储介质承载有一个或多个计算机程序,所述一个或多个计算机程序能够被所述电子设备执行,以使所述电子设备实现如上所述的单细胞分析方法。
54、从上述的技术方案可以看出,本技术公开了一种单细胞分析方法、装置、电子设备和存储介质,该方法和装置应用于电子设备,具体为基于scatac方法对目标细胞进行分析,得到目标细胞的第一数据;基于scrna方法对目标细胞进行分析,得到目标细胞的第二数据;基于scatac方法和scrna方法对第一数据和第二数据进行整合分析,得到目标细胞的细胞类型和可及性。本方案通过关联和注释方法对单细胞的染色质开发区域进行精确分析,从而能够为染色质可及性的研究提供数据基础。
55、另外,本技术的机身方案还具有如下的技术效果:
56、1、通过整理encode数据库中人和小鼠两个物种已知的启动子、增强子、沉默子和绝缘子的注释信息,对于其他物种这根据基因注释的gtf文件,将peaks分为启动子、远端调控元件和intergenic peak,将基因组上一块区域的位置的数字信息,关联上了其可能调控的基因信息,实现了对已有的细胞类型marker库的资源、geneontology、kegg pathway等功能信息的充分利用,进一步实现了从类间差异可及性peaks中对启动子相关peaks的筛选,启动子的可及性对基因转录表达的影响最直接,方便用户快速的关注到marker基因启动子在不同细胞群中的特异分布情况,提高了scatac细胞类型注释的效率,节省用户的分析时间。
57、2.、本技术中genescore算法不是对基因上下游100kb范围peaks的可及性数据的简单累加,充分考虑了表观调控对基因表达的经典模式,启动子区域对基因的表达影响最大,随着peak到tss距离的增加,peak对基因表达影响的作用逐渐减小,采用变化的权重模型,将peaks的可及性转化为了gene水平的可及性得分。对于大量公开的scrna单细胞数据,或者用户实验设计中的单细胞转录组数据,这部分数据都是基因维度的表达数据,将peaks的可及性数据转化为基因维度的genescore后,为scatac和scrna的数据整合,建立scrna-scatac的细胞映射关系,进行scatac的细胞类型注释提供了数据支持,同时提高了scrna数据的利用率。
58、3、scatac多样本合并支持根据基因组位置10kb为一个bin生成tilemartix,首先cellranger-atac分析软件对于每个样本都会生成一套10~15万个peaks,样本间peaks的数目以及生物学上相同的peak可能在起始和终止位置不同的情况,非常不利于多样本的数据整合,采用tilematirx后每个样本根据基因组和染色体长度划分的bin数目是固定的,就可以直接采用seurat的lsi对样本降维,然后cca整合多个样本,整合后的数据进行第一次聚类,然后对聚类结果的每个群的细胞单独进行peaks calling,最后合并全部细胞群的peaks,这个方法的好处时,预分群将染色体可及性模式相近的细胞聚为一群,对每个群的细胞单独callpeaks,可以尽可能地保留细胞群(或细胞类型)特异的peaks。基于合并后的peaks重新计算每个样本在peaks上的可及性矩阵,然后再进行合并和降维聚类分析时,保证了各个样本的peaks维度的统一。
59、4、除了本技术中的scatac-scrna的一一映射方法外,也有直接采用scatacgenescore与scrna基因表达直接计算两两细胞相关性的方法,矩阵中包含的基因在2万多的水平,同时由于数据的稀疏性问题,采用pearson相关的方法评估scatac和scrna细胞间的相似性存在大家都在0.8以上,细胞间相关性的差异不明显,本方法基于scrna的pca降维结果和scatac的lsi降维结果进行cca映射到一个标准化的二维空间内,有效解决了高维空间距离算法的缺陷。
60、5、pseudobulk的方法中不管是基于拟时间分析结果还是亚群的分析结果,对于peaks、genescore、tf-motif和基因表达数据,都采用50个细胞合并成一个metacell也称为pseudobulk,首先使得scrna 70~80%和scatac~90%的数据稀疏性降维了40%作用,为后续进行表观联合分析提供了数据稳定性保证,另外合并的50个细胞的数据均产自客观的检测数据,避免了magic算法、saver、dca、scimpute和alra等软件可能带来的数据填充的偏好性问题。
61、6、只有在发育分化进程中表达变化的基因才是真正参与调控作用基因,首先先选择随拟时间变化最大的2000个基因,一下就从3万个基因的研究范围缩小到~7%左右,进一步筛选genescore与基因表达模式显著相关的基因,进一步锁定受表观调控的基因,更深层次的挖掘peak-基因表达显著相关的关系对,挖掘出对特定仅影响最大的调控元件peak,进一步结合tf-tfmotif的共表达分析筛选出参与调控的转录因子,最终再结合peaks和tf的结合关系,选择与tf结合peak且peak与基因表达相关性有显著升高的分子调节路径。这种递进化的表观调控机制研究方法,逻辑严密,且逐步缩小研究的目标和范围,提高了分析结果的有效性,同时极大的减少了分析的所需的时间和计算资源,缩短项目的研究周期。
- 还没有人留言评论。精彩留言会获得点赞!