无细胞DNA分析中基因融合检测的方法和应用与流程
无细胞dna分析中基因融合检测的方法和应用
1.本技术是申请日为2016年10月10日,申请号为201680071449.4,发明名称为“无细胞dna分析中基因融合检测的方法和应用”的申请的分案申请。
2.交叉引用
3.本技术要求于2015年10月10日提交的美国临时申请号62/239,879的权益,该申请通过引用以其整体并入本文。
4.发明背景
5.癌性细胞可以具有融合到一起的染色体。如果对这样的染色体进行测序,它将生成可以被映射到基因组的两个不同区域(在相同或不同染色体上)的读段。基因融合在基因结构的进化中起作用。重复、序列趋异和重组是基因进化工作的主要参与者。当基因融合在非编码序列区域中发生时,它可以导致目前在另一个基因的顺式调节序列的控制下的基因的表达的错误调节。如果它在编码序列中发生,基因融合可以引起新基因的装配,允许通过在多结构域蛋白中增加肽模块而出现新功能。
6.染色体显带分析(chromosome banding analysis)、荧光原位杂交(fish)和逆转录聚合酶链式反应(rt-pcr)是在诊断实验室使用的常见方法。由于癌症基因组的非常复杂的性质,这些方法都具有其独特的缺点。近期开发诸如高通量测序和定制dna微阵列具有引进更高效的方法的希望,但仍然不足。高通量基因组测序技术已经被用作研究工具并且目前正在被引入临床,而在个性化医学的未来,全基因组测序数据可能是指导治疗干预的重要工具。
7.发明概述
8.在一个方面,公开了用于通过以下来确定基因融合的系统和方法:确定含有融合的染色体dna分子的至少一部分的测序数据的融合读段;确定基因组上的预定点,其中融合读段的至少一个映射部分在预定点(断点)处剪切(clipped);将来自两个断点(断点对)的两个映射的读段部分识别为潜在的融合候选;基于断点对创建一个或更多个融合集并将融合集聚类到一个或更多个融合簇中;和将符合预定标准的每个融合簇识别为基因融合。
9.在一个方面,本公开内容提供了用于处理来自样品的遗传序列读段数据的方法,所述方法包括:确定含有融合的染色体dna分子的至少一部分的测序数据的融合读段;确定基因组上的预定点,其中融合读段的至少一个映射部分在所述预定点(断点)处剪切;将来自两个断点(断点对)的两个映射的读段部分识别为潜在的融合候选;基于断点对创建一个或更多个融合集并将融合集聚类到一个或更多个融合簇中;和将符合预定标准的每个融合簇识别为基因融合。
10.在一些实施方案中,所述方法包括向每个读段分配独特的分子或读段标识符(读段id)。在一些实施方案中,所述方法包括从一侧或两侧剪切所述读段的每个映射部分。在一些实施方案中,断点的身份独立于所述读段并且由符号、染色体和位置来标识。在一些实施方案中,断点记录统计数据,所述统计数据包括在所述断点处被剪切或分裂的读段和分子的数目,以及跳过所述断点的野生型读段和分子的数目。在一些实施方案中,所述方法包括选择具有公共读段id的每两个映射读段部分作为潜在融合候选,所述公共读段id属于具
有适当符号的两个断点。在一些实施方案中,映射之前的原始读段中的潜在融合候选位置将所述读段部分显示为原先彼此相邻地定位。在一些实施方案中,所述方法包括检查读段部分是否映射在一条链上以获得断点符号的差异。在一些实施方案中,所述方法包括追踪融合集统计数据。
11.在一些实施方案中,所述融合集统计数据是断点id、包含在所述集中的分子或读段的数目。在一些实施方案中,所述方法包括将具有相似断点的融合集分组为融合簇。在一些实施方案中,所述相似断点是相距不超过5个核苷酸、不超过10个核苷酸或不超过25个核苷酸的断点。在一些实施方案中,所述方法包括定义基因组中两个区域之间的融合簇。在一些实施方案中,所述方法包括为所述融合簇确定每个配偶体的融合分子的数目。在一些实施方案中,所述方法包括为所述融合簇确定每个配偶体的融合读段的数目。在一些实施方案中,所述方法包括为所述融合簇确定每个配偶体的野生型分子的数目。在一些实施方案中,所述方法包括为所述融合簇确定每个配偶体的野生型读段或分子的数目。在一些实施方案中,所述方法包括为所述融合簇确定每个配偶体的融合百分比为融合分子与每个配偶体的总分子的比率。在一些实施方案中,总分子包括野生型和剪切组分。在一些实施方案中,所述方法包括为所述融合簇确定每个配偶体的基因信息。在一些实施方案中,所述方法包括确定所述融合簇的下游基因。在一些实施方案中,所述标准包括在所述簇中具有多于一个分子或者具有至少一个具有沃森和克里克链两者的分子。
12.在一个方面,本公开内容提供了分析遗传信息的系统,包括:dna测序仪;处理器,所述处理器偶联至所述dna测序仪,所述处理器运行处理来自样品的遗传序列读段数据的计算机代码,所述计算机代码包括用于以下的指令:确定含有融合的染色体dna分子的一部分的测序数据的融合读段;确定基因组上的至少一个预定点,其中融合读段的至少一个映射部分在所述预定点(断点)处剪切;将来自两个断点(断点对)的两个映射的读段部分识别为潜在的融合候选;基于断点对创建一个或更多个融合集并将融合集聚类到一个或更多个融合簇中;和将符合预定标准的每个融合簇识别为基因融合。
13.在一个方面,本公开内容提供了包括以下的方法:用dna测序仪对dna分子进行测序以生成序列的集合;将序列的集合映射到参考基因组;从映射的集合识别融合读段,其中融合读段包含子序列,其中第一子序列映射至第一遗传基因座且第二子序列映射到第二、独特的遗传基因座;对于每个融合读段,识别第一遗传基因座处的第一断点和第二遗传基因座处的第二断点,其中断点是融合读段的序列被剪切的参考基因组上的点,且其中第一断点和第二断点形成断点对;生成融合读段集,每个集包含具有相同断点对的融合读段;聚类融合读段集,其中每个簇由具有第一预定多核苷酸距离内的第一断点和第二预定核苷酸距离内的第二断点的融合读段集形成;和确定一个或更多个簇的基因融合,其中簇的基因融合具有选自簇中的第一断点的断点作为第一融合基因断点和选自簇中的第二断点的断点作为第二融合基因断点,并且其中第一融合基因断点和第二融合基因断点各自基于选择标准选择。
14.在一些实施方案中,独特的遗传基因座位于不同染色体上或位于同一染色体的不同基因上。在一些实施方案中,第一预定距离和第二预定距离各自不超过5个核苷酸、不超过10个核苷酸或不超过25个核苷酸。在一些实施方案中,选择标准包括断点在簇中具有最多融合读段。在一些实施方案中,所述方法包括确定多个基因簇的基因融合。
15.在一个方面,本公开内容提供了包括以下的方法:用dna测序仪对多个dna分子进行测序;用标识符为多个序列分子的每一个加标签;将每个加标签的序列映射到参考基因组;从映射的加标签的序列识别剪切读段,其中剪切读段是包含映射部分和剪切部分的加标签的序列,其中映射部分映射到遗传基因座且剪切部分不映射到遗传基因座;确定每个剪切读段的断点,其中断点是剪切读段的序列被剪切的参考基因组上的点;生成断点集,每个断点集包括具有相同断点的剪切读段的标识符;通过比较断点集的对创建断点对的集,断点对的每个集包括存在于比较的断点集的对的两个成员中的标识符;聚类断点对的集,其中每个簇包括具有第一预定遗传距离内的对的第一断点和第二预定遗传距离内的对的第二断点的断点对的集;和确定一个或更多个簇的基因融合,其中簇的基因融合具有选自簇中的第一断点的断点作为第一融合基因断点和选自簇中的第二断点的断点作为第二融合基因断点,并且其中第一融合基因断点和第二融合基因断点各自基于选择标准选择。在一些实施方案中,选择标准包括断点在簇中具有最多融合读段。
16.在一个方面,本公开内容提供了用于识别融合基因断点的方法,所述方法包括:确定含有融合的染色体dna分子的至少一部分的测序数据的融合读段;确定基因组上的预定点,其中融合读段的至少一个映射部分在预定点(断点)处剪切;将来自两个断点(断点对)的两个映射的读段部分识别为潜在的融合候选;基于断点对创建一个或更多个融合集并将融合集聚类到一个或更多个融合簇中;将符合预定标准的每个融合簇识别为基因融合,并将基因融合的断点识别为融合基因断点。
17.在一个方面,本公开内容提供了用于诊断受试者中的状况的方法,所述方法包括:确定含有融合的染色体dna分子的至少一部分的测序数据的融合读段;确定基因组上的预定点,其中融合读段的至少一个映射部分在预定点(断点)处剪切;将来自两个断点(断点对)的两个映射的读段部分识别为潜在的融合候选;基于断点对创建一个或更多个融合集并将融合集聚类到一个或更多个融合簇中;和将符合预定标准的每个融合簇识别为基因融合,其中所述基因融合指示该状况。
18.在一些实施方案中,状况是癌症。在一些实施方案中,癌症选自由血液学癌症、肉瘤和前列腺癌组成的组。在一些实施方案中,所述方法还包括向受试者施用治疗。
19.通过引用并入
20.本说明书中提及的所有出版物、专利和专利申请通过引用并入本文,其程度如同每一个单独的出版物、专利或专利申请被具体和单独地指明通过引用并入的相同程度。
21.附图简述
22.本发明的新颖特征在所附权利要求中具体地阐述。通过参考下文的详细描述和附图将获得对本发明的特征和优点的更好理解,详细描述阐述了利用本发明的原理的说明性实施例,在附图中:
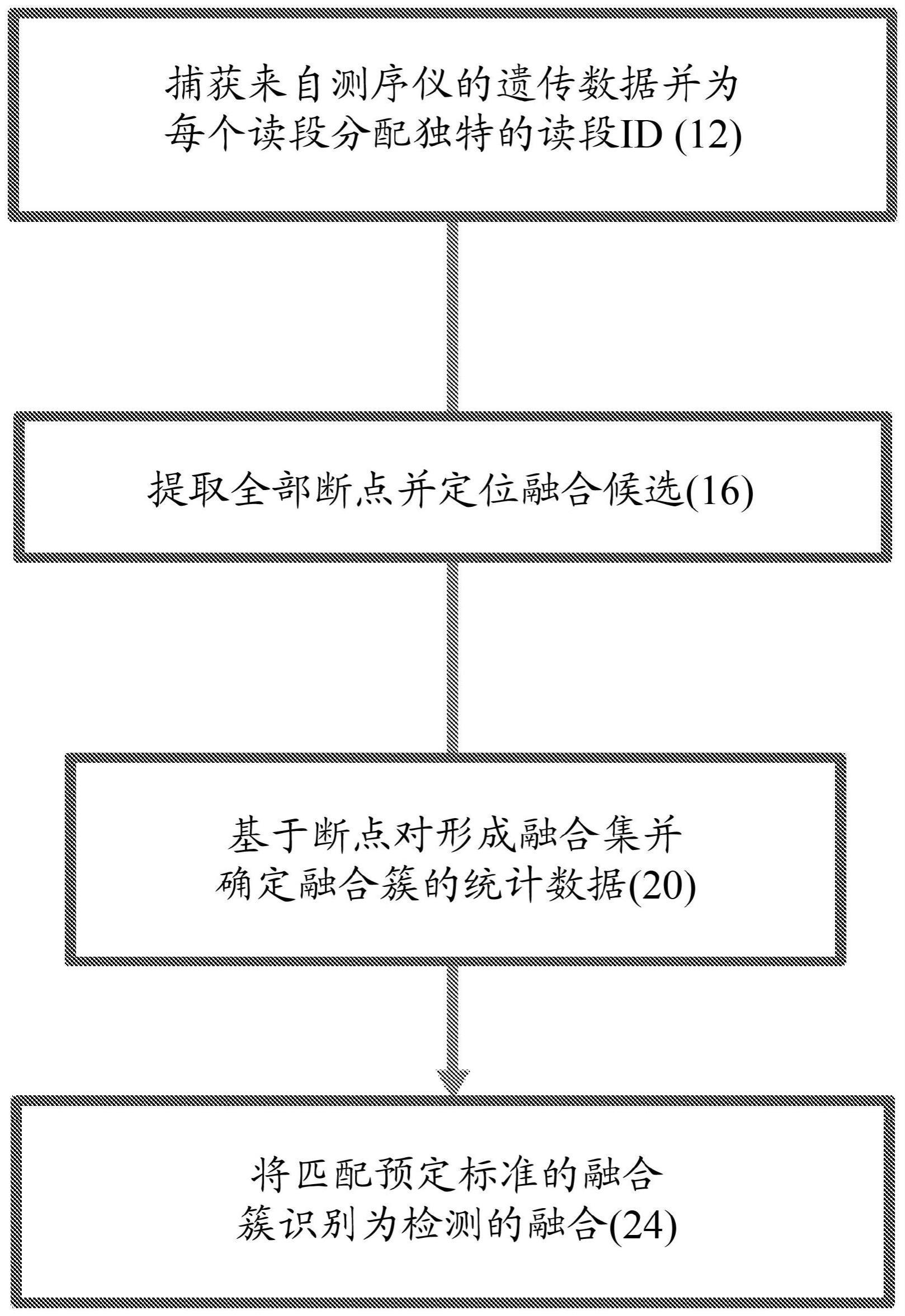
23.图1示意了用于检测基因融合的示例性过程。
24.图2描绘了从两个其他染色体产生融合染色体的可能的不同情景。
25.图3a示出了示例性+/-断点和分别从左侧/右侧被剪切的读段部分。
26.图3b示出了基因融合检测使用的示例性合并过程。
27.图4示意了本发明的系统。
28.图5示出了染色体a和染色体b之间的示例性基因融合,以及跨断点映射的dna融合
读段。
29.图6示出了dna片段到参考基因组中的两个位置的示例性映射。
30.图7示出了其中映射的断点被不同地定位的示例性融合读段。
31.图8示出了映射的融合读段示例性分组到集中并将集分组成簇,用于判定融合基因断点。
32.图9a-9c示出了基因融合检测过程的示例性图解。
33.发明的详细描述
34.本发明涉及用于检测基因融合的系统和方法。
35.融合基因的断点的准确映射是具有挑战性的。当试图映射断点时,测序中的错误和比对融合基因的困难仅仅是遇到的两个困难。本文描述的系统和方法可以提供以下优势中的一个或更多个。系统可以识别可能导致肿瘤形成的融合基因,因为融合基因可以比非融合基因产生活跃得多的异常的蛋白。当融合基因是引起癌症的致癌基因时,系统准确地确定癌症存在;这些包括bcr-abl、tel-aml1(具有t(12;21)的all)、aml1-eto(具有t(8;21)的m2 aml)、和在染色体21上具有中间缺失的tmprss2-erg,通常发生于前列腺癌中。在tmprss2-erg的情况下,通过用致癌基因ets转录因子破坏雄激素受体(ar)信号通路和抑制ar表达,融合产物调节前列腺癌。多数融合基因从血液学癌症、肉瘤和前列腺癌发现。致癌融合基因可以导致具有新功能或不同于两个融合配偶体的功能的基因产物。可选地,将原致癌基因融合至强启动子,并且从而致癌基因功能通过由上游融合配偶体的强启动子引起的上调发挥功能。后者常见于淋巴瘤,其中致癌基因与免疫球蛋白基因的启动子并列。致癌基因融合转录物还可以由反式剪接或连读事件引起。从基因组序列和结构的角度分析这些基因融合可以提供引导开发改进的癌症诊断和靶向疗法的相关数据。
36.图1示出了用于确定基因融合的示例性过程。一般来说,该过程捕获来自测序仪的遗传数据并且应用比对和连接测序的对-末端读段的融合技术,该对-末端读段的插入尺寸小于两个读段长度之和,并且在融合后为每个读段分配独特的读段标识符(读段id)12。接下来,该过程提取所有断点并定位融合候选16。随后,该过程基于断点对形成融合集并确定融合簇的统计数据20。随后,通过匹配预定标准将融合簇识别为检测的融合24。
37.下面讨论关于图1的过程的细节。癌性细胞可以具有融合到一起的染色体。如果对这样的染色体进行测序,它将生成可以被映射到基因组的两个不同区域(在相同或不同染色体上)的读段。利用此行为来检测融合。
38.映射前,独特的读段标识符(读段id)被分配给每个读段并将在一个或更多个fastq文件中的读段标题(header)中编码,如以下详述的。可选地,独特分子,诸如包含独特条形码的寡核苷酸,可以代替读段id使用。一旦fastq文件被映射,此编码的读段id将被检索并且它可以容易地示出何种匹配来自相同原始读段。随后,该过程提取所有断点:融合读段(含有来自融合染色体的部分dna分子的测序数据的读段)不能以整体映射到基因组并且映射器将其不同的部分映射到基因组上的不同位置。当使用传统技术来尝试断点映射时,这呈现了挑战。此类读段的每个映射部分从一侧或两侧被剪切。断点是融合读段的至少一个映射部分被剪切的基因组上的点。断点的身份独立于所述读段并且由其符号、染色体和位置来标识。+/-断点具有分别从左侧/右侧被剪切的读段部分。从一个位置的同一侧剪切或分裂的全部读段列在相关的断点读段列表中。断点还可以记录其他统计数据,如在所述
断点处被剪切或分裂的读段和分子的数目;或者跳过断点的野生型读段和分子的数目。还可以提供断点位置处的基因信息。通过分配断点,使用或不使用聚类,本文描述的方法和系统可被用于准确地确定发生基因融合的基因座。
39.随后,此过程发现融合:具有公共读段id的每两个映射读段部分是潜在的融合候选,所述公共读段id属于具有适当符号的两个断点。它们还需要具有表明读段部分最初彼此相邻地定位的正确的片段顺序(它们在映射之前在原始读段中的位置),以被认为是真正的融合候选。此外,所得的融合必须在序列链方面是生物学上可能的。这仅意味着,如果读段部分被映射到相同链上(都在5’链或都在3’链),则断点的符号必须不一致,且反之亦然。在图2中示出这个的实例。
40.所有提取的融合候选基于断点对放置在融合集中。融合集还可以记录统计数据,如断点id和分子的数目以及包含在所述集中的读段。这些统计数据可以被追踪。
41.随后进行聚类。具有足够靠近的断点的所有融合集将分组在一个融合簇中。结果是,融合簇被定义为在基因组的两个区域之间。本公开内容还提供了对于融合簇,确定每个配偶体的融合分子的数目、每个配偶体的融合读段的数目、每个配偶体的多个野生型分子、每个配偶体的野生型读段或分子的数目、或者每个配偶体的融合百分比作为融合分子相对于每个配偶体的总分子的比率。
42.图5示出了染色体a和染色体b之间的假设性基因融合。作为交换的结果,基因融合包含每个染色体的一部分。交换的点被称为断点。在无细胞dna中,dna片段可以跨断点映射,诸如融合读段1、融合读段2和融合读段3。
43.测序产生dna片段的序列。软件用识别标签标记每个序列。软件还将这些产生的序列映射到参考基因组上。图6示出了融合读段1向参考基因组的假设性映射。映射软件将融合读段的序列映射到发现足够同源性的参考基因组中的任何位置。有歧义的序列可以被映射到参考基因组中的多个位置。
44.在融合读段跨融合基因的断点映射的情况中,软件通常将融合读段的序列映射两次,向每个染色体映射一次。但是,在每个情况中,映射软件不能适当地将序列的部分(子序列)映射到参考基因组。因此,映射序列将包括映射到参考基因组的子序列和由于同源性差而未映射到同一基因座的子序列二者。此类子序列被称为“剪切”序列。读段被剪切处的参考基因组上的点是断点。
45.由于每个序列携带识别标签,映射到两个不同位置的序列可以由于相同标签被鉴定为源自相同原始序列。所以,例如,具有足够同源性的序列的子序列被映射到染色体a,且具有不足同源性的序列的子序列被剪切。类似地,映射软件将序列映射到染色体b,在存在不足同源性处剪切序列。
46.然而,作为几个因素,包括测序中的错误和映射算法的特征的结果,包括融合基因的断点的dna片段可能不会准确地映射到每个参考染色体上的断点基因座。例如,映射软件可能识别实际断点稍微上游或者稍微下游的序列的断点。这些错误可以影响例如取决于准确的基因融合信息的癌症诊断的准确性。
47.几个假设的映射错误在图7中示出。融合读段1适当地映射,断点在参考染色体中被指示为断点a1和断点b1(第一断点和第二断点)。此融合读段具有断点对a1-b1。融合读段2不适当地映射,染色体1的断点被确定为在上游,在断点a2(第一断点)处。然而,染色体b中
的断点被正确映射在断点b1(第二断点)处。此融合读段具有断点对a2-b1。融合读段3也不适当地映射,染色体a的断点在断点a1(第一断点)处正确地映射,但染色体b的断点被确定为在下游,在断点b2(第二断点)处。此融合读段具有断点对a1-b2。在此情况下,软件已经识别了融合基因的几个断点。
48.根据本公开内容的方法,为了判定融合基因中的断点,映射序列基于共有断点对被分组成集,并随后基于参考基因组中的预定碱基距离内的断点被分组成簇。
49.这种方法在图8中描述。融合读段1、2、3、4、5和6的序列被映射到参考基因组的染色体a和b。其中断点在融合的两侧的序列被分组成集。在此实例中,融合读段1和4共有断点对a1和b1,并且被分组到集i。融合读段2和5共有断点对a2和b1,并且被分组到集ii。融合读段3和6共有断点对a1和b2,并且被分组到集iii。
50.在此实例中,断点a1和a2在预定遗传距离a(例如10个碱基)内,并且断点b1和b2在预定遗传距离b内。因此,集i、ii和iii被分组到一个簇内。
51.使用用户选择的选择标准判定融合基因断点。在一些实施方案中,标准包括在所述簇中具有多于一个分子和/或者具有至少一个具有沃森和克里克链两者的分子。在一个方法中,每个染色体中的断点由投票方法确定,其中所有断点中具有最多相关融合读段的断点被判定为融合基因断点。在其他方法中,不同序列的断点可以使用质量算法来加权。在图8的实例中,在染色体a中,断点a1与4个融合读段相关,而断点a2与2个融合读段相关。因此,第一基因融合断点被判定为在a1处。在染色体b中,断点b1与4个融合读段相关,而断点b2与2个融合读段相关。因此,第二基因融合断点被判定为在b1处。
52.另一个示例性方法在图9a-9c中示出。dna分子使用dna测序系统测序,诸如高通量测序仪。序列可以被分析以生成集合中的原始分子的共有序列。生成的序列的集合用独特的标识符加标签(在此情况下,1-7)。序列被映射到参考基因组。在此实例中,序列各自映射到参考基因组中的两个不同位置。映射部分被描绘为短棒,而剪切部分被描绘为虚线。所有序列的断点被识别。在此实例中,染色体a上的断点是a1、a2和a3。染色体b上的断点是b1、b2和b3。映射读段基于共有断点被组织成集。断点对被确定为具有相同标识符和每个染色体上的相同断点的序列的对。确定染色体与簇断点上的预定距离。在此实例中,簇包括断点a1和a2以及b1和b2。断点a3和b3在预定距离以外,并且因此不被包括在簇中。基于选择标准判定原始分子中的断点对。在此实例中,标准是基于投票。因此,断点a1和b1基于具有在簇内的多数分子而被判定为断点对。
53.系统可以处理剪接中常见的短序列的多个比对。更长的序列可以在融合点周围获得以确认其比对。为了降低假阳性率,可以使用一系列过滤器,包括读段数目、序列相似性、序列位置分布过滤器以提供具有高特异性的结果。另外,可以使用表达估计工具rsem(通过期望最大化的rna-seq,rna-seq by expectation maximization)来应用具有稀疏优化的期望最大化(em)算法来估计幻想的转录物丰度。此外,此丰度定量可以增加识别准确性。综合考虑,这些特征允许系统提供基因融合事件的更完整的综述。
54.系统可以包括以任何合适的文件格式从一个或更多个样品获得多个序列读段、识别重复序列读段集和对于每个重复序列读段集仅存储一个读段。合适的文件格式包括fasta和fastq文件格式。fasta和fastq是用于存储来自高通量测序的原始序列读段的常用文件格式。fastq文件存储每个序列读段的标识符、序列和每个读段的质量评分串。fasta文
件仅存储标识符和序列。这两种文件格式是许多常见测序比对和装配算法的输入。本发明认识到,样品内和跨样品的fastq和fasta文件的读段序列信息倾向于高度冗余或重复。这意味着,许多序列读段将由相同序列组成。本发明的方法利用此冗余性来实现文件大小的多倍减小,并且在存储数据的检索方面没有损失。例如,本发明可以用于读取与样品相关的fasta/fastq文件并且在主读段序列文件中仅存储独特的读段序列。
55.系统还包括为与识别的独特序列具有相同序列的每个读段收集元信息(meta information),诸如读段标识符。然后可以将该元信息写入该样品的文件中,其中元信息与原始fasta/fastq文件中识别的独特序列读段相关联,并且现在存储在主读段序列文件中。由于这个新文件不包含原始文件中发现的重复信息,它比原始文件更小并且更容易传输。而且,压缩文件根本不需要包含任何实际的序列数据。在某些方面,压缩文件可以简单地包含索引到存储在主文件中的独特序列的序列读段的标识符。
56.序列数据可以通过获得——使用包括偶联到非瞬时性存储器的处理器的计算机系统——多个序列读段来压缩。每个序列读段可以包括序列串以及元信息。序列读段可以以一个或更多个fasta或fastq文件的格式提供,例如,元信息包括描述行(前面带有“》”字符),并且任选地在fastq的情况下包括质量评分。序列串优选地表示核苷酸序列数据,例如使用iupac核苷酸编码。识别仅包含独特条目的序列串的子集。然后可以使用本发明的系统和方法来写入包括所识别的子集和——对于多个序列读段的每一个——用该子集中表示该序列读段的独特条目的指示符的序列读段的元信息的输出。
57.在一些实施方案中,子集(即仅包含独特序列读段)被写入主读段文件,其可以是文本文件。优选地,使用iupac核苷酸编码在主读段文件中表示独特序列读段,使得文件是人类可读的并且进一步处理可以容易地执行(例如使用脚本语言诸如perl或python)。元信息可写入对应于输入fasta或fastq文件的压缩输出文件。
58.方法可以包括仅从输出重构原始输入,并且在某些实施方案中,检索是无损的,甚至是完全无损的。也就是说,输出可以被处理以创建包含多个序列读段的新fasta或fastq文件。在检索是无损的情况下,新fasta或fastq文件包含与fasta或fastq文件相同的信息。
59.本发明适用于任何合适类型的数据文件。除了前面提到的fasta和fastq文件,序列读段还可以在变体判别格式(variant call format,vcf)文件中捕获。随着高通量测序的进展,多个测序中心检测人类基因组中的变体并通过这些vcf文件报告它们是常见的。本发明可以促进统一数据库的开发,以便在不同来源的vcf文件中存储变体信息,从而使研究人员跨中心执行复杂的等位基因、样本和群体水平的查询。统一数据库可以通过在一个通用等位基因表上存储每个独特等位基因(例如,独特序列读段)并通过将这些独特等位基因的参考存储到关联样品和样品水平元数据来整合来自不同样品的vcf文件中的变体信息。
60.系统的实现可以包括用于压缩序列数据的方法。该方法包括:使用包括偶联到非瞬时性存储器的处理器的计算机系统获得多个序列读段,每个序列读段包括序列串和元信息;识别仅包含独特条目的序列串的子集;写入包括所述子集和——对于所述多个序列读段中的每一个——用该子集中代表该序列读段的独特条目的指示符的序列读段的元信息的输出。优选地,输出包括使用iupac核苷酸编码存储子集的一个或更多个文本文件。优选地,输出被存储为纯文本(例如,并且可以使用文本编辑器程序打开并由人在屏幕上阅读)。在优选实施方案中,序列读段无损地存储。该方法可以包括处理输出以创建包含多个序列
读段的新fasta或fastq文件。多个序列读段可以作为fasta或fastq文件获得,且新fasta或fastq文件可以包含与fasta或fastq文件相同的信息。在一些实施方案中,输出占用小于存储获得的多个序列读段所需的磁盘空间的%。
61.现在将描述用于获得样品、生成测序读段和用于实施本发明的各种类型的测序的一般方法。应该理解,这些示例性方法不是限制性的,并且可以根据需要由本领域技术人员进行修改。
62.获得多个序列读段可以包括对来自样品的核酸进行测序以产生序列读段。如下文详细解释的,获得多个序列读段还可以包括接收来自测序仪的测序数据。样品中的核酸可以是任何核酸,包括例如组织样品中的基因组dna、从实验室样品中特定靶扩增的cdna或来自多个生物体的混合dna。在一些实施方案中,样品包括来自单倍体或二倍体生物体的纯合dna。例如,样品可以包括来自罕见隐性等位基因纯合的患者的基因组dna。在其他实施方案中,样品包括来自具有体细胞突变使得两种相关核酸以除了50%或100%的等位基因频率,即20%、5%、1%、0.1%或任何其他等位基因频率存在的二倍体或多倍体生物体的杂合遗传物质。
63.在一个实施方案中,从含有多种其他组分(诸如蛋白、脂质和非模板核酸)的生物样品分离核酸模板分子(例如dna或rna)。核酸模板分子可以从获自动物、植物、细菌、真菌或任何其他细胞生物体的任何细胞材料获得。用于在本发明中使用的生物样品还包括病毒颗粒或制备物。核酸模板分子可直接从生物体或从生物体获得的生物样品获得,例如从血液、血清、血浆、尿液、脑脊液、唾液、粪便、淋巴液、滑液、囊液、腹水、胸膜渗出物、羊水、绒毛膜绒毛样品、来自植入前胚胎的液体、胎盘样品、灌洗液和宫颈阴道液、组织间隙液、颊拭子样品、痰、支气管灌洗液、巴氏涂片样品(pap smear sample)或眼液。任何组织或体液样本(例如体液样本的人类组织)可以用作在本发明中使用的核酸的来源。核酸模板分子也可以从培养的细胞诸如原代细胞培养物或细胞系分离。从其获得模板核酸的细胞或组织可以被病毒或其他细胞内病原体感染。样品也可以是从生物样本中提取的总rna、cdna文库、病毒或基因组dna。样品也可以是来自非细胞来源的分离的dna。
64.从生物样品获得的核酸可以被片段化以产生合适的片段用于分析。模板核酸可以使用各种机械、化学和/或酶学方法片段化或修剪(sheared)成期望的长度。dna可以经由使用例如由covaris(woburn,mass)销售的超声波仪超声处理、短暂暴露于dna酶、或使用一种或更多种限制酶的混合物、或转座酶或切口酶随机修剪。rna可以通过短暂暴露于rna酶、加热加上镁或通过修剪来片段化。rna可以被转化成cdna。如果使用片段化,则rna可以在片段化之前或之后转化成cdna。在一个实施方案中,核酸通过超声片段化。在另一个实施方案中,核酸通过液压剪床(hydroshear)仪器来片段化。通常,单个核酸模板分子可以是从约2kb碱基至约40kb。在特定的实施方案中,核酸是约6kb-10kb片段。核酸分子可以是单链、双链或具有单链区的双链(例如,茎和环结构)。
65.生物样品可以根据需要在洗涤剂或表面活性剂的存在下裂解、均质化或分级。合适的洗涤剂可以包括离子型洗涤剂(例如十二烷基硫酸钠或n-月桂酰肌氨酸)或非离子型洗涤剂。一旦核酸从样品中提取或分离出来,就可以被扩增。
66.扩增是指产生核酸序列的另外的拷贝,并且通常使用聚合酶链式反应(pcr)或本领域已知的其他技术进行。扩增反应可以是扩增核酸分子的本领域已知的任何扩增反应诸
如pcr。其他扩增反应包括巢式pcr、pcr-单链构象多态性、连接酶链式反应、链置换扩增和限制性片段长度多态性、基于转录的扩增系统、滚环扩增、和超分枝滚环扩增、定量pcr、定量荧光pcr(qf-pcr)、多重荧光pcr(mf-pcr)、实时pcr(rtpcr)、限制性片段长度多态性pcr(pcr-rflp)、原位滚环扩增(rca)、桥式pcr、皮升pcr(picotiter pcr)、乳液pcr、转录扩增、自我维持序列复制、共有序列引物pcr、任意引物pcr、简并寡核苷酸引物pcr和基于核酸的序列扩增(nabsa)。可以使用的扩增方法包括在美国专利第5,242,794号;第5,494,810号;第4,988,617号;和第6,582,938号中描述的那些。在某些实施方案中,扩增反应为例如美国专利第4,683,195号;以及美国专利第4,683,202号中描述的pcr,特此通过引用并入。用于pcr、测序和其他方法的引物可以通过克隆、直接化学合成和本领域已知的其他方法来制备。引物也可以从商业来源获得,诸如eurofins mwg operon(huntsville,ala.)或life technologies(carlsbad,calif.)。
67.扩增衔接子可以附接到片段化的核酸上。衔接子可以是商购获得的,例如来自integrated dna technologies(coralville,iowa)。在某些实施方案中,衔接子序列用酶附接到模板核酸分子。酶可以是连接酶或聚合酶。连接酶可以是能够将寡核苷酸(rna或dna)连接至模板核酸分子的任何酶。合适的连接酶包括可从new england biolabs(ipswich,mass.)商购可得的t4 dna连接酶和t4 rna连接酶。使用连接酶的方法是本领域熟知的。聚合酶可以是能够将核苷酸添加到模板核酸分子的3'和5'末端的任何酶。
68.连接可以是平端的或利用互补的悬垂末端。在某些实施方案中,片段的末端可以在片段化之后被修复、修剪(trimmed)(例如使用核酸外切酶)、或填充(例如使用聚合酶和dntp)以形成平端。在一些实施方案中,使用商业试剂盒(诸如从epicenter biotechnologies(madison,wis.)可获得的试剂盒)进行末端修复以生成平端5'磷酸化核酸末端。在生成平端时,末端可以用聚合酶和datp处理以形成独立添加至片段的3'末端和5'末端的模板,从而产生单个a悬垂。这个单个a用于指导被称为t-a克隆的方法中从5'末端的单个t悬垂的片段的连接。可选地,因为限制性消化后限制酶留下的悬垂的可能的组合是已知的,末端可以保持原样,即不齐的末端。在某些实施方案中,使用具有互补悬垂末端的双链寡核苷酸。
69.本发明的实施方案涉及将条形码序列附接至模板核酸。在某些实施方案中,条形码被附接到每个片段。在其他实施方案中,多个条形码(例如两个条形码)被附接到每个片段。条形码序列通常包括使序列在测序反应中有用的某些特征。例如,条形码序列被设计为在条形码序列内具有最小均聚物区域或者不具有均聚物区域,所述均聚物区域即在一行具有2个或更多个相同碱基,诸如aa或ccc。条形码序列还被设计为使得它们在执行逐个碱基测序时距离碱基添加顺序至少有一个编辑距离,确保第一个碱基和最后一个碱基与序列的预期碱基不匹配。
70.条形码序列被设计为使得每个序列与核酸的特定部分相关,允许序列读段与它们来自于其的部分相关联。在某些实施方案中,条形码序列的范围从约5个核苷酸至约15个核苷酸。在特定实施方案中,条形码序列的范围从约4个核苷酸至约7个核苷酸。由于条形码序列与模板核酸一起测序,寡核苷酸长度应该是最小的长度,以便允许附接来自模板核酸的最长读段。例如,多个dna条形码可以包含各个数目的核苷酸序列。在某些实施方案中,条形码序列包含2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、
27、28、29、30或更多个核苷酸。当仅附接到多核苷酸的一个末端时,多个dna条形码可以产生2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30或更多个不同的标识符。可选地,当附接到多核苷酸的两个末端时,多个dna条形码可以产生4、9、16、25、36、49、64、81、100、121、144、169、196、225、256、289、324、361、400或更多个不同的标识符(当dna条形码仅附接到多核苷酸的一个末端时为^2)。
71.通常,条形码序列与模板核酸分子间隔至少一个碱基(使均聚物组合最小化)。在某些实施方案中,条形码序列用例如酶附接到模板核酸分子。如下所述,酶可以是连接酶或聚合酶。
72.扩增或测序衔接子或条形码或其组合可以附接到片段化的核酸上。这样的分子可以商购获得,例如从integrated dna technologies(coralville,iowa)。在某些实施方案中,这样的序列用酶诸如连接酶附接至模板核酸分子。合适的连接酶包括可从new england biolabs(ipswich,mass.)商购可得的t4 dna连接酶和t4 rna连接酶。连接可以平端的或者通过使用互补的悬垂末端。在某些实施方案中,在片段化之后,片段的末端可以被修复、修整(例如使用核酸外切酶)或填充(例如使用聚合酶和dntp)以形成平端。在一些实施方案中,使用商业试剂盒(诸如从epicenter biotechnologies(madison,wis.)可获得的试剂盒)进行末端修复以生成平端5'磷酸化核酸末端。在生成平端时,末端可以用聚合酶和datp处理以形成独立添加至片段的3'末端和5'末端的模板,从而产生单个a悬垂。这个单个a可以指导被称为t-a克隆的方法中从5'末端的单个t悬垂的片段的连接。可选地,因为限制性消化后限制酶留下的悬垂的可能的组合是已知的,末端可以保持原样,即不齐的末端。在某些实施方案中,使用具有互补悬垂末端的双链寡核苷酸。
73.在任何处理步骤(例如获得、分离、片段化、扩增或加条形码)后,可以对核酸进行测序。
74.测序可以是通过本领域已知的任何方法。dna测序技术包括使用标记的终止子或引物和在平板或毛细管中的凝胶分离的经典双脱氧测序反应(sanger法)、使用可逆终止的标记的核苷酸的合成测序、焦磷酸测序、454测序、illumina/solexa测序、与标记的寡核苷酸探针的文库的等位基因特异性杂交、使用与标记的克隆的文库的等位基因特异性杂交然后连接的合成测序、实时监测聚合步骤期间标记的核苷酸的掺入、群落测序(polony sequencing)、solid测序靶向测序、单分子实时测序、外显子测序、基于电子显微术的测序、小组测序(panel sequencing)、晶体管介导的测序、直接测序、随机鸟枪测序、全基因组测序、杂交测序、毛细管电泳、凝胶电泳、双链测序、循环测序、单碱基延伸测序、固相测序、高通量测序、大规模平行签名测序(massively parallel signature sequencing)、乳液pcr、低变性温度共扩增-pcr(cold-pcr)、多重pcr、可逆染料终止子测序、配对的末端测序、近末端测序(near-term sequencing)、核酸外切酶测序、连接测序、短读段测序、单分子测序、实时测序、反向终止子测序、纳米孔测序、ms-pet测序及其组合。在一些实施方案中,测序方法可以是大规模平行测序,即,同时(或以快速相继)测序至少100、1000、10,000、100,000、1百万、1千万、1亿、或10亿个多核苷酸分子的任一个。在一些实施方案中,测序可以通过基因分析仪进行,诸如例如可以从illumina或applied biosystems商业获得的基因分析仪。最近已经通过使用聚合酶或连接酶的连续或单一延伸反应以及通过与探针文库的单一或顺序差异杂交证明了分离的分子的测序。测序可以通过dna测序仪(例如设计用于执行测序反应
的机器)来执行。
75.可以使用的测序技术包括例如使用合成测序系统。在第一步中,将dna修剪成大约300-800个碱基对的片段,并将片段平端化。然后将寡核苷酸衔接子连接到片段的末端。衔接子充当用于片段的扩增和测序的引物。片段可以使用例如含有5'-生物素标签的衔接子b附接至dna捕获珠,例如链霉亲和素包被的珠。附接于珠的片段在油-水乳液的液滴内进行pcr扩增。结果是每个珠上克隆扩增的dna片段的多个拷贝。在第二步中,珠被捕获在孔中(皮升大小)。焦磷酸测序在每个dna片段上平行进行。添加一个或更多个核苷酸生成由测序仪器中的ccd照相机记录的光信号。信号强度与掺入的核苷酸数目成正比。焦磷酸测序利用了在添加核苷酸时释放的焦磷酸(ppi)。ppi在腺苷5’磷酰硫酸的存在下被atp硫酸化酶转化为atp。萤光素酶使用atp将萤光素转化为氧化萤光素,并且该反应生成被检测和分析的光。
76.可以使用的dna测序技术的另一个实例是来自life technologies corporation(carlsbad,calif.)的applied biosystems的solid技术。在solid测序中,将基因组dna修剪成片段,并将衔接子附接到片段的5'和3'末端以生成片段文库。可选地,内部衔接子可以通过将衔接子连接到片段的5'和3'末端、使片段环化、消化环化的片段以生成内部衔接子而被引入,并且将衔接子附接到所得片段的5'和3'末端来生成匹配配对文库。接下来,在含有珠、引物、模板和pcr组分的微量反应器中制备克隆珠群体。pcr后,将模板变性并富集珠以分离具有延伸模板的珠。所选的珠上的模板经受3'修饰,这允许键合到玻璃载玻片。该序列可通过部分随机寡核苷酸与由特定荧光团识别的中心确定碱基(或碱基对)的连续杂交和连接来确定。记录颜色后,去除连接的寡核苷酸,且然后重复该过程。
77.可以使用的dna测序技术的另一个实例是离子半导体测序,使用例如life technologies(south san francisco,calif.)的ion torrent的以商标ion torrent出售的系统。离子半导体测序在例如rothberg等人,an integrated semiconductor device enabling non-optical genome sequencing,nature 475:348-352(2011);美国公布2010/0304982;美国公布2010/0301398;美国公布2010/0300895;美国公布2010/0300559;和美国公布2009/0026082中描述,其每一个的内容通过引用整体并入。
78.可以使用的测序技术的另一个实例是illumina测序。illumina测序是基于使用折回pcr(fold-back pcr)和锚定引物在固体表面上扩增dna。基因组dna被片段化,且衔接子被添加到片段的5'和3'末端。附着于流通池通道的表面的dna片段被延伸并被桥式扩增。片段变成双链的,且双链分子被变性。固相扩增随后变性的多个循环可在流通池的每个通道中产生相同模板的约1,000个拷贝的单链dna分子的几百万个簇。使用引物、dna聚合酶和四种荧光团标记的可逆终止核苷酸进行顺序测序。掺入核苷酸后,使用激光激发荧光团并捕获图像并记录第一个碱基的身份。来自每个掺入的碱基的3'终止子和荧光团被去除并且重复掺入、检测和鉴定步骤。根据此技术的测序在美国专利第7,960,120号;美国专利第7,835,871号;美国专利第7,232,656号;美国专利第7,598,035号;美国专利第6,911,345号;美国专利第6,833,246号;美国专利第6,828,100号;美国专利第6,306,597号;美国专利第6,210,891号;美国公布2011/0009278;美国公布2007/0114362;美国公布2006/0292611;和美国公布2006/0024681中描述,其每一个通过引用整体并入。
79.可以使用的测序技术的另一个实例包括pacific biosciences(menlo park,
calif.)的单分子实时(smrt)技术。在smrt中,四种dna碱基中的每一种都与四种不同荧光染料中的一种附接。这些染料是磷酸连接的。单个dna聚合酶与模板单链dna的单个分子固定在零模波导(zmw)的底部。将一个核苷酸掺入到增长链需要几毫秒。在此期间,荧光标签被激发并产生荧光信号,且荧光标签被裂解。染料的相应荧光的检测指示掺入了哪个碱基。该过程重复进行。
80.可以使用的测序技术的另一个例子是纳米孔测序(soni&meller,2007,progress toward ultrafast dna sequence using solid-state nanopores,clin chem 53(11):1996-2001)。纳米孔是直径为1纳米的级别的小孔。由于离子通过纳米孔的传导,将纳米孔浸没在导电流体中并在其上施加电势产生轻微的电流。流动的电流的量对纳米孔的尺寸敏感。当dna分子穿过纳米孔时,dna分子上的每个核苷酸以不同程度阻碍纳米孔。因此,当dna分子穿过纳米孔时流经纳米孔的电流的变化代表dna序列的读取。
81.可以使用的测序技术的另一个实例包括使用化学敏感的场效应晶体管(chemfet)阵列对dna进行测序(例如,如美国公布2009/0026082中所述的)。在该技术的一个实例中,dna分子可以被置于反应室中,并可将模板分子与结合聚合酶的测序引物杂交。在测序引物的3'末端将一个或更多个三磷酸掺入新核酸链可以通过chemfet的电流变化来检测。阵列可以具有多个chemfet传感器。在另一个实例中,单核酸可以附接到珠上,并且核酸可以在珠上扩增,并且单个珠可以被转移到chemfet阵列上的单个反应室中,每个室具有chemfet传感器,并且核酸可以被测序。
82.可以使用的测序技术的另一个实例包括使用电子显微镜,如例如由moudrianakis,e.n.和beer m.在base sequence determination in nucleic acids with the electron microscope,iii.chemistry and microscopy of guanine-labeled dna,pnas 53:564-71(1965)中描述的。在该技术的一个实例中,单个dna分子使用可用电子显微镜区分的金属标记物来标记。然后将这些分子在平坦表面上拉伸并使用电子显微镜成像以测量序列。
83.根据本发明的实施方案的测序生成多个读段。根据本发明的读段通常包括长度小于约150个碱基或长度小于约90个碱基的核苷酸数据的序列。在某些实施方案中,读段在约80和约90个碱基之间,例如约85个碱基长度。在一些实施方案中,本发明的方法应用于非常短即长度小于约50或约30个碱基的读段。序列读段数据可以包括序列数据以及元信息。如本领域技术人员已知的,序列读段数据可以以任何合适的文件格式存储,包括例如vcf文件、fasta文件或fastq文件。
84.fasta最初是用于搜索序列数据库的计算机程序,并且名称fasta也指标准文件格式。参见pearson&lipman,1988,improved tools for biological sequence comparison,pnas 85:2444-2448。fasta格式的序列以单行描述开始,接着是序列数据行。描述行通过第一列中的大于(“》”)符号与序列数据区分开。“》”符号后面的单词是序列的标识符,且该行的其余部分是描述(都是任选的)。“》”和标识符的第一个字母之间不应有空格。建议文本的所有行少于80个字符。如果出现以“》”开头的另一行,则序列结束;这表示另一个序列的开始。
85.fastq格式是用于存储生物序列(通常是核苷酸序列)及其相应的质量评分的基于文本的格式。它与fasta格式相似,但是在序列数据之后具有质量评分。为简洁起见,序列字
母和质量评分都使用单个ascii字符编码。fastq格式是用于存储高通量测序仪器(诸如illumina genome analyzer)的输出的实际标准。cock等人,2009,the sanger fastq file format for sequences with quality scores,and the solexa/illumina fastq variants,nucleic acids res 38(6):1767-1771。
86.对于fasta和fastq文件,元信息包括描述行而不是序列数据行。在一些实施方案中,对于fastq文件,元信息包括质量评分。对于fasta和fastq文件,序列数据在描述行之后开始,并且通常使用一些iupac模糊代码的子集(任选地带有
“‑”
)呈现。在优选实施方案中,序列数据将使用a、t、c、g和n字符,任选地根据需要包括
“‑”
或者包括u(例如,以表示空位或尿嘧啶)。
87.在一些实施方案中,至少一个主序列读段文件和输出文件被存储为纯文本文件(例如使用诸如ascii;iso/iec 646;ebcdic;utf-8或utf-16的编码)。本发明提供的计算机系统可以包括能够打开纯文本文件的文本编辑器程序。文本编辑器程序可以指能够在计算机屏幕上呈现文本文件(诸如纯文本文件)的内容、允许人员编辑文本(诸如使用监视器、键盘和鼠标)的计算机程序。示例性文本编辑器包括但不限于microsoft word、emacs、pico、vi、bbedit和textwrangler。优选地,文本编辑器程序能够在计算机屏幕上显示纯文本文件,以人类可读格式显示元信息和序列读段(例如,不是二进制编码而是使用字母数字字符,因为它们将用于打印人类书写)。
88.虽然已经参照fasta或fastq文件讨论了方法,但是本发明的方法和系统可以用于压缩任何合适的序列文件格式,包括例如variant call format(vcf)格式的文件。典型的vcf文件将包括标题部分和数据部分。标题包含任意数量的元信息行,每行都以字符
‘
##’开始,以及以单个
‘
#’字符开始的tab分隔字段定义行。字段定义行命名八个必填列,而主体部分包含填充字段定义行定义的列的数据行。vcf格式在danecek等人,2011,the variant call format and vcftools,bioinformatics 27(15):2156-2158中描述。标题部分可被视为要写入压缩文件的元信息,并且数据部分可被视为行,其中每一行只有在独特时才被存储在主文件中。
89.本发明的某些实施方案提供了序列读段的装配。例如,在通过比对进行装配时,读段彼此比对或与参考比对。通过比对每个读段,继而与参考基因组比对,所有读段关于彼此定位以创建装配体。另外,将序列读段与参考序列比对或映射到参考序列也可以用于识别序列读段中的变体序列。识别变体序列可以与本文描述的方法和系统组合使用,以进一步帮助疾病或状况的诊断或预后或用于指导治疗决定。
90.在一些实施方案中,本发明的任何或全部步骤是自动化的。可选地,本发明的方法可以全部或部分地在一个或更多个专用程序中实现,例如每一个任选地以编译语言诸如c++写入,然后以二进制编译和分发。本发明的方法可以全部或部分地作为现有序列分析平台内的模块或通过调用现有序列分析平台内的功能而实现。在某些实施方案中,本发明的方法包括响应于单个启动队列(例如,源自人类活动、另一个计算机程序或机器的触发事件中的一个或组合)而自动调用的多个步骤。因此,本发明提供了其中任何步骤或步骤的任何组合可以响应于队列而自动发生的方法。自动地通常意味着不介入人类输入、影响或交互(即仅响应于原始或预先排队的人类活动)。
91.该系统还包括各种形式的输出,其包括主题核酸的准确和灵敏的解释。检索的输
出可以以计算机文件的格式提供。在某些实施方案中,输出是fasta文件、fastq文件或vcf文件。输出可被处理以产生文本文件或含有序列数据诸如与参考基因组的序列比对的核酸的序列的xml文件。在其他实施方案中,处理产生包含坐标或描述主题核酸中相对于参考基因组的一个或更多个突变的串的输出。本领域中已知的比对串包括简单无空位比对报告(simple ungapped alignment report,sugar)、详细的有用的标记空位比对报告(verbose useful labeled gapped alignment report,vulgar)和简要比对信息表达式(compact idiosyncratic gapped alignment report,cigar)(ning,z.等人,genome research 11(10):1725-9(2001))。这些串例如在来自european bioinformatics institute(hinxton,uk)的exonerate序列比对软件中实现。
92.在一些实施方案中,产生包含cigar串的序列比对——诸如,例如序列比对图(sam)或二元比对图(bam)文件(sam格式在例如li等人,the sequence alignment/map format and samtools,bioinformatics,2009,25(16):2078-9中描述)。在一些实施方案中,cigar显示或包括每行一个空位比对。cigar是一种报告为cigar串的压缩的成对比对格式。cigar串对于表示长(例如基因组)成对比对是有用的。在sam格式中使用cigar串来表示读段与参考基因组序列的比对。
93.cigar串遵循建立的基序。每个字符前面是数字,给出事件的碱基计数。使用的字符可以包括m、i、d、n和s(m=匹配;i=插入;d=缺失;n=空位;s=置换)。cigar串定义匹配/不匹配和缺失(或空位)的序列。例如,cigar串2md3m2d2m将意味着,比对包含2个匹配、1个缺失(为了节省一些空间省略数字1)、3个匹配、2个缺失和2个匹配。
94.如本发明所预期的,上文描述的功能可以使用包括软件、硬件、固件、硬接线或者这些的任何组合的本发明的系统来实现。实现功能的特征还可以物理地位于各个位置,包括被分布为使得功能的各部分在不同的物理位置处实现。
95.如本领域技术人员将认识到必要或最适合于执行本发明的方法,本发明的计算机系统或机器包括一个或更多个处理器(例如中央处理单元(cpu)、图形处理单元(gpu)或二者)、主存储器和静态存储器,其经由总线彼此通信。
96.图4图示了适于执行本发明的方法的系统701。如图4中示出的,系统701可以包括服务器计算机705、终端715、测序仪725、测序仪计算机721、计算机749中的一个或更多个,或其任何组合。每个这样的计算机设备可以经由网络709通信。测序仪725可以任选地包括或可操作地偶联到其自己的、例如专用测序仪计算机721(包括任何输入/输出机构(i/o)、处理器和存储器)。另外地或可选地,测序仪725可以经由网络709可操作地偶联到服务器705或计算机749(例如,膝上型计算机、台式计算机或平板电脑)。计算机749包括一个或更多个处理器、存储器和i/o。在本发明的方法采用客户机/服务器结构的情况下,本发明的方法的任何步骤可以使用服务器705来执行,该服务器705包括能够获得数据、指令等的处理器、存储器和i/o中的一个或更多个,或者通过接口模块提供结果或将结果作为文件提供。服务器705可以通过计算机749或终端715接合网络709,或者服务器705可以直接连接到终端715。终端715优选地是计算机设备。根据本发明的计算机优选地包括偶联到i/o机构和存储器的一个或更多个处理器。
97.处理器可以由一个或更多个处理器提供,包括例如单核或多核处理器中的一个或更多个。i/o机构可以包括视频显示单元(例如,液晶显示器(lcd)或阴极射线管(crt))、字
母数字输入设备(例如键盘)、光标控制设备(例如鼠标)、磁盘驱动单元、信号发生设备(例如扬声器)、加速度计、麦克风、蜂窝射频天线以及网络接口设备(例如网络接口卡(nic)、wi-fi卡、蜂窝调制解调器、数据插孔、以太网端口、调制解调器插孔、hdmi端口、迷你hdmi端口、usb端口)、触摸屏(例如crt、lcd、led、amoled、超级amoled)、指示设备、触控板、光(例如led)、光/图像投影设备或其组合。根据本发明的存储器指的是由一个或更多个有形设备提供的非瞬时性存储器,所述有形设备优选地包括在其上存储一组或更多组指令的一个或更多个机器可读介质(例如软件),所述指令实现本文描述的方法或功能中的任何一个或更多个。软件还可以在被系统701内的计算机执行期间完全或至少部分地驻留在主存储器、处理器或二者中,主存储器和处理器也构成机器可读介质。软件还可以经由网络接口设备通过网络发送或接收。
98.尽管机器可读介质可以在示例性实施方案中被示为是单个介质,但是术语“机器可读介质”应该被认为包括存储一组或多组指令的单个介质或多个介质(例如,集中式或分布式数据库、和/或相关联的高速缓存和服务器)。术语“机器可读介质”还应当包括能够储存、编码或携带指令集的任何介质,其中这些指令集由机器执行或使机器执行本发明的任何一种或更多种方法。存储器可以是例如硬盘驱动器、固态驱动器(ssd)、光盘、闪存、压缩盘、磁带驱动器、“云”存储位置中的一个或更多个,或其组合。在某些实施方案中,本发明的设备包括用于存储器的有形的、非瞬时性计算机可读介质。用作存储器的示例性装置包括半导体存储器装置(例如eprom、eeprom、固态驱动器(ssd)和闪存装置例如sd、微sd、sdxc、sdio、sdhc卡);磁盘(例如内部硬盘或可移动磁盘);和光盘(例如cd和dvd盘)。
99.在一些实施方案中,本公开内容的方法和系统可以用于诊断疾病或状况,例如癌症。如本文使用的,术语“诊断”是指技术人员能够估计和/或确定患者是否患有给定疾病或状况的方法。在一些实施方案中,本发明的方法可以用于疾病或状况(例如癌症)的疾病的预后。如本文使用的,术语“预后”是指疾病或状况进展的可能性,包括疾病或状况的复发。在一些实施方案中,本发明的方法可用于评估发展疾病或状况(例如癌症)的风险。例如,本文描述的方法和系统可用于鉴定与特定诊断、预后或者发展疾病或状况的风险相关的断点或基因融合。此外,本文所述的方法和系统可用于鉴定与预测的治疗结果相关的断点或基因融合。因此,所述方法和系统可以用于指导疾病或状况的治疗(例如通过向受试者施用化合物或剂)或指导用于治疗疾病或状况的药物的制备。
100.如本文使用的,“治疗”疾病或状况是指采取步骤以获得有益或期望的结果,包括临床结果。有益或期望的临床结果包括但不限于缓解或改善与疾病或状况相关的一种或更多种症状。如本文使用的,向受试者“施用(administering)”或“施用(administration of)”化合物或剂可以使用本领域技术人员已知的多种方法中的一种进行。例如,化合物或剂可以静脉内、动脉内、皮内、肌肉内、腹膜内、静脉内、皮下、眼部、舌下、经口(通过摄入)、鼻内(通过吸入)、脊椎内、脑内和透皮(通过吸收,例如通过皮肤导管)施用。化合物或剂也可以通过可再充装或生物可降解的聚合物装置或其他装置(例如贴剂和泵)或制剂适当地引入,所述装置或制剂提供化合物或剂的延长、缓慢或控制释放。施用还可以进行例如一次、多次和/或在一个或更多个延长的时间段内进行。在某些方面,施用包括直接施用(包括自我施用)和间接施用(包括给药物开处方的行为)。例如,如本文使用的,指导患者自我施用药物或者由另一个人施用药物和/或向患者提供药物处方的医师正在向患者施用药物。
在一些实施方案中,化合物或剂通过口服例如通过摄入施用至受试者,或者经静脉地例如通过注射施用至患者。在一些实施方案中,口服施用的化合物或剂处于延长释放或缓慢释放制剂中,或者使用用于这种缓慢或延长释放的装置施用。
101.如本文使用的,术语“癌症”包括但不限于各种类型的恶性赘生物,其中大多数可侵入周围组织,并可以转移至不同部位(参见例如pdr medical dictionary,第1版(1995),其出于所有目的通过引用整体并入本文)。术语“赘生物(neoplasm)”和“肿瘤(tumor)”是指通过细胞增殖比正常组织更迅速地生长并且在去除引发增殖的刺激之后继续生长的异常组织。这种异常组织显示部分或完全缺乏结构组织和与可以是良性(例如良性肿瘤)或恶性(例如恶性肿瘤)的正常组织的功能协调。癌症的一般类别的实例包括但不限于上皮癌(carcinoma)(源自上皮细胞的恶性肿瘤诸如例如乳腺癌、前列腺癌、肺癌和结肠癌的常见形式)、肉瘤(源自结缔组织或间充质细胞的恶性肿瘤)、淋巴瘤(源自造血细胞的恶性肿瘤)、白血病(源自造血细胞的恶性肿瘤)和生殖细胞肿瘤(源自全能性细胞的肿瘤,在成人中最常见于睾丸或卵巢中;在胎儿、婴儿和年幼儿童中,最常见于身体中线,特别是在尾骨尖处)、急变性肿瘤(类似未成熟组织或胚胎组织的典型恶性肿瘤)等。意图被本发明涵盖的赘生物类型的实例包括但不限于与神经组织、血液形成组织、乳房、皮肤、骨骼、前列腺、卵巢、子宫、子宫颈、肝、肺、脑、喉、胆囊、胰腺、直肠、甲状旁腺、甲状腺、肾上腺、免疫系统、头和颈、结肠、胃、支气管和/或肾的癌症相关的那些赘生物。在特定实施方案中,可以被检测的癌症的类型和数目包括但不限于血癌、脑癌、肺癌、皮肤癌、鼻癌、喉癌、肝癌、骨癌、淋巴瘤、胰腺癌、皮肤癌、肠癌、直肠癌、甲状腺癌、膀胱癌、肾癌、口腔癌、胃癌、实体瘤(solid state tumors)、异质肿瘤、均质肿瘤等。在特定实施方案中,癌症是血液学癌症、肉瘤或前列腺癌。
102.虽然本文已经显示和描述了本发明的优选实施方案,但对于本领域技术人员将明显的是,此类实施方案仅通过示例的方式提供。在不偏离本发明的情况下,本领域技术人员现在将想到许多变化、改变和替换。应当理解,在实践本发明时可以采用本文描述的本发明的实施方案的各种替代选择。所附权利要求意图界定本发明的范围,并且从而涵盖在这些权利要求范围内的方法和结构及其等同物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1