基于NLRs信号通路对泛癌患者预后相关基因的免疫浸润分析及预后模型构建的方法和应用
本发明属于医药,尤其涉及一种基于nlrs信号通路对泛癌患者预后相关基因的免疫浸润分析及预后模型构建的方法和应用。
背景技术:
1、目前,各种癌症的治疗手段包括手术切除、放疗、化疗和靶向治疗等,但仍然存在许多治疗上的挑战。
2、nod样受体(nlrs)在检测多种病原体和启动先天免疫反应中起着关键作用,它驱动nfkb和mapk的激活,导致细胞因子和细胞凋亡的产生。此外,一些模式识别受体(如nlrp1和nlrp3)的nlr寡聚导致多蛋白复合物的形成,导致caspase-1激活,然后il-1β和il-18炎症细胞因子的出现诱导细胞死亡,与乳腺癌、胃癌、肺癌和皮肤癌的侵袭性生长有关。鉴于nlrs在免疫反应和炎症调节中的关键作用,了解nlr信号通路如何影响癌症至关重要。近几年,有相关研究显示乳腺癌、胰腺腺癌、gbm的kegg功能分析表明,其癌症相关基因主要富集在nod样受体信号通路中。然而,目前对于nlrs在泛癌中的作用及其信号通路机制的认识还很有限。因此,有必要对nlrs进一步探索,为泛癌的治疗和预后评估提供新的思路和方法。
技术实现思路
1、本发明要解决的技术问题是提供一种基于nlrs信号通路对泛癌患者预后相关基因的免疫浸润分析及预后模型构建的方法和应用。
2、为解决上述技术问题,本发明采用以下技术方案:
3、基于nlrs信号通路对泛癌患者预后相关基因的免疫浸润分析及预后模型构建的方法,通过分析基于tcga数据库中泛癌的基因表达数据和临床数据,筛选出差异表达基因;对其基因进行单因素和多因素cox回归分析得到构建预后模型的最佳基因,绘制高低风险组之间的生存曲线;为了评估预后预测模型风险得分是否受其他临床因素影响,进行独立预后分析,并构建roc曲线验证模型的准确性;此外,还进行高低风险组之间的免疫细胞浸润分析。
4、上述方法,包括以下步骤:
5、(1)从kegg数据库(https://www.genome.jp/kegg/)下载nod样受体通路相关的所有基因(共收集到184个基因);
6、(2)对tcga数据库中blca、brca、coad、esca、gbm、hnsc、luad、lusc、read、stad的基因表达数据进行差异分析,得到对应癌症的正常样本和患病样本的差异表达基因;
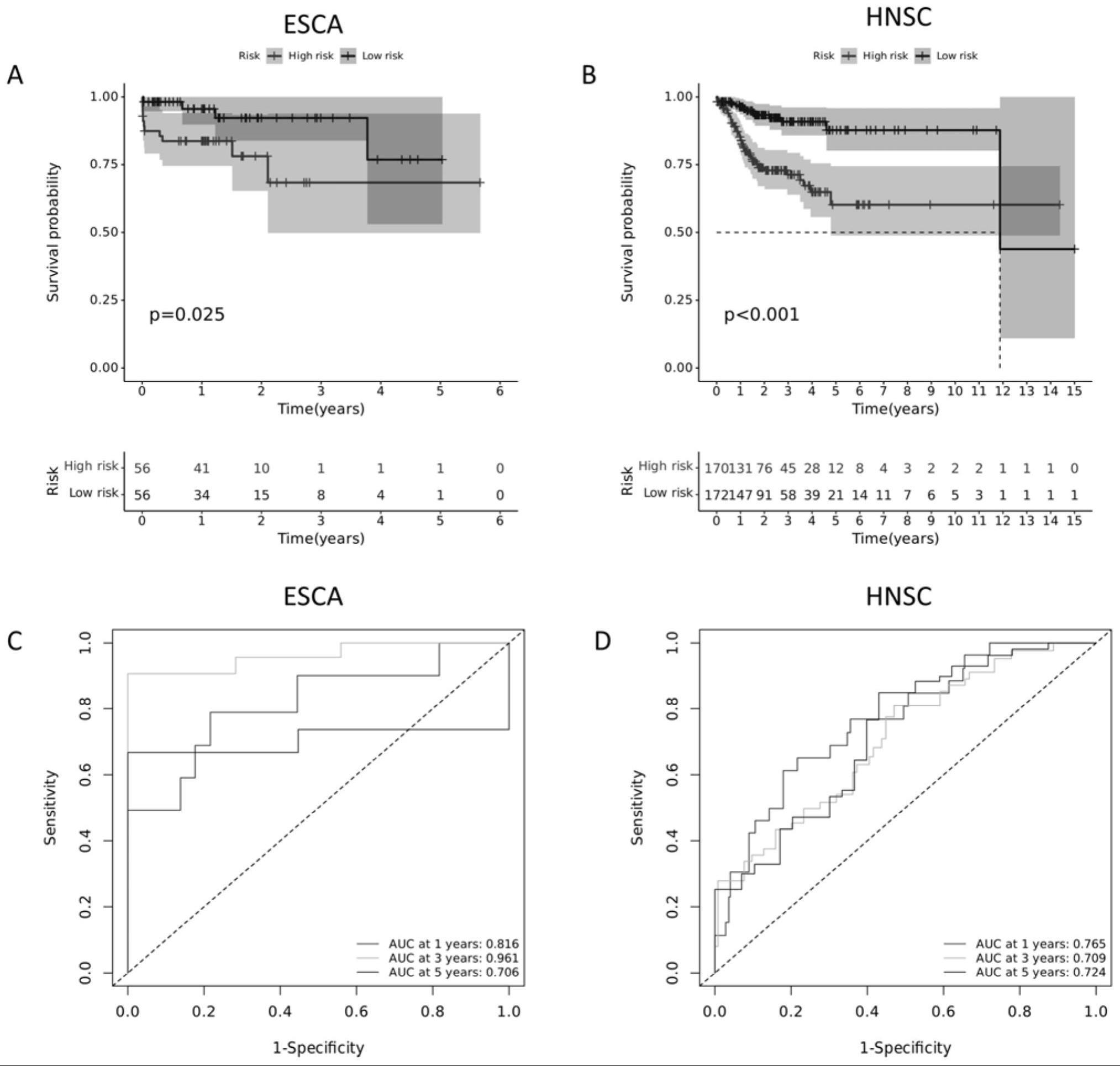
7、(3)将差异表达基因进行单因素cox回归分析,得到与泛癌预后相关基因,再对其基因进行多因素cox回归分析,筛选出构建预后预测模型的最佳基因;根据风险评分公式,计算每个样本的风险得分,按照中位风险评分,将样本分为高风险组和低风险组,绘制高低风险组之间的生存曲线;
8、(4)为了评估预后预测模型的风险得分是否受其他临床因素影响,对风险评分和临床因素进行独立预后分析,并绘制roc曲线验证预后预测模型的准确性;
9、(5)对高低风险组进行免疫细胞浸润分析,得到在高低风险组中浸润水平存在差异的免疫细胞。
10、步骤(2)中:
11、从tcga(https://www.cancer.gov/ccg/research/genome-sequencing/tcga)下载10种癌症的正常样本和患病样本的基因表达矩阵以及对应的临床数据,使用"edger"r包筛选正常和肿瘤样本之间的差异表达基因,筛选标准为|log2 fold change(fc)|>1且错误发现率(fdr)<0.05;临床数据包括患者的性别、年龄、生存状态、病理分期、生存期等信息。
12、步骤(3)中:对差异基因进行单因素cox回归分析,评估各基因与患者生存的关系,计算相应的风险比(hr)和95%置信区间(hr95h、hr95l)的上下限;基于这些结果,选择具有显著预后价值的基因进行多元回归分析,筛选出构建预后模型的最佳基因;根据风险评分公式,计算出每个样本的风险评分,将所有样本的风险评分从小到大进行排序,以风险得分中值为临界值,将样本分为高表达组或低表达组;为了研究风险评分与总生存率之间的关系,采用“survival”软件包对每种癌症类型进行log-rank检验,且p值小于0.05被认为具有统计学意义;生存分析采用kaplan-meier方法。
13、步骤(4)中:在独立预后分析中,风险评分作为预测变量,其他临床因素(包括患者年龄、性别、临床分期)作为协变量;对风险评分和临床因素进行通过计算风险评分的风险比(hazard ratio,hr)和95%置信区间的上限和下限(hr95h,hr95l),评估风险评分在考虑其他因素的情况下对患者生存期的预测能力;使用survival、survminer和timeroc软件包生成随时间变化的受试者工作特征(roc)曲线。用曲线下面积(auc)来证明预测的准确性,auc值越高,对应的roc曲线越靠近左上角,表示真阳性率越高,假阳性率越低。也就是说,auc值越大,表示预测精度越高。
14、步骤(5)中:为了估计基因表达与8种免疫浸润细胞丰度的相关性,使用r包“gsva”采用ssgsea算法;具体而言,通过多变量cox回归分析获得的风险评分将样本分为高表达组和低表达组,并使用ssgsea算法评估nod样受体相关基因表达与免疫浸润细胞丰度的相关性;8种免疫浸润细胞包括:activated cd4 t cell,activated dendritic cell,effectormemory cd8 t cell,gamma delta t cell,immature b cell,natural killer cell,neutrophil,plasmacytoid dendritic cell。
15、上述方法用于预测泛癌患者预后生存时间。
16、上述方法在建立预测泛癌1年、2年、3年生存概率的nomogram模型中的应用。
17、nomogram模型的输入因素为风险评分,性别,年龄和病理分期。nomogram图根据模型中选择的多个临床因素对最终结果变量的影响程度,对每个影响因素的取值范围进行评分。在预测单个样本时,可以将每个影响因素的得分相加,得到总分。然后将总分与结果事件概率之间的关系用于诊断或预测疾病的发生和进展,提供个体结果事件的预测价值。
18、针对目前泛癌的治疗和预后评估存在的问题,发明人建立了一种基于nlrs信号通路对泛癌患者预后相关基因的免疫浸润分析及预后模型构建的方法,通过分析基于tcga数据库中泛癌的基因表达数据和临床数据,筛选出差异表达基因;对其基因进行单因素和多因素cox回归分析得到构建预后模型的最佳基因,绘制高低风险组之间的生存曲线;为了评估预后预测模型风险得分是否受其他临床因素影响,进行独立预后分析,并构建roc曲线验证模型的准确性。根据预后模型构建列线图,预测患者生存时间,具有重要的临床参考意义。此外,还进行高低风险组之间的免疫细胞浸润分析,提高对泛癌免疫治疗的预测反应。
19、综上,本发明通过生物信息学整合分析,深入研究了预后基因及免疫细胞对泛癌的作用机制,评估nlrs相关基因对泛癌的预后价值;且本预后预测模型较其他模型准确率更高,为泛癌的诊断和预防,药物靶标治疗提供了新的研究思路,探索可能作为药物靶标的免疫细胞来提高患者的生存率。
- 还没有人留言评论。精彩留言会获得点赞!