一种对肿瘤RNA测序数据的病毒检测方法及系统
本发明涉及一种基于多模态的深度学习方法,用于肿瘤数据的病毒识别,属于健康大数据处理。
背景技术:
1、全世界所有肿瘤病例中大约15%是由病毒产生的。国际肿瘤研究署(iarc)已经明确将11种病原体分类为人类致癌剂,其中包括多种病毒。这些病毒能够通过多种机制促使正常细胞变为恶性肿瘤,从而加剧肿瘤的发展。病毒相关的肿瘤通常可以追溯到病毒与宿主细胞之间的复杂相互作用。某些病毒可以直接感染宿主细胞,破坏宿主细胞的dna或rna,导致异常细胞增殖。例如,人乳头瘤病毒(hpv)感染与宫颈癌的关联就是一个经典的例子。hpv感染可以引发宫颈上皮细胞的异常增生,最终导致宫颈癌的发展。
2、随着下一代测序技术的快速发展,可以帮助我们更深入地研究病毒与肿瘤之间的联系。这些测序技术的问世使我们能够更全面、更迅速地识别新的病毒株。通常,研究人员会基于已知病毒的序列信息,通过比对大规模的dna或rna测序数据,来寻找与已知病毒高度相似的序列,从而识别新的病毒株。
3、目前,对肿瘤测序数据进行病毒鉴定的现有方法主要分为两类。第一类是基于比对的方法,其主要原理是将测序读数与已知病毒基因组序列进行比对,以鉴别病毒的存在。然而,这类方法存在以下问题:比对效率较低,尤其在面对大规模数据时,计算成本较高;同时,现有病毒数据库的覆盖范围有限,难以发现新的病毒种类,因此在病毒鉴定方面存在局限性。
4、第二类方法是基于免比对的方法,这些方法借助机器学习技术,通过学习序列的特征信息对病毒进行分类,从而实现病毒的识别。然而,现存的免比对方法存在精度较低的问题,特别是在处理复杂的肿瘤组学数据时,往往难以实现准确的病毒组成识别。现有方法通常没有针对肿瘤组学数据的优化,因此在这一领域的应用受到了限制。
5、现有技术在肿瘤测序数据中识别病毒方面存在一系列问题,包括耗时较长、效率低、精度不高等诸多不足之处。这些问题限制了有效识别肿瘤测序数据中的病毒,从而妨碍了相关研究和临床应用的进展。
6、因此,提出一种有效识别肿瘤测序数据中的病毒的方法是十分有意义的。
技术实现思路
1、为应对上述问题,本发明提供了一种基于多模态深度学习的方法,用于识别肿瘤测序数据中的病毒。
2、根据本发明的一方面,提出了一种对肿瘤rna测序数据的病毒检测方法,包括:
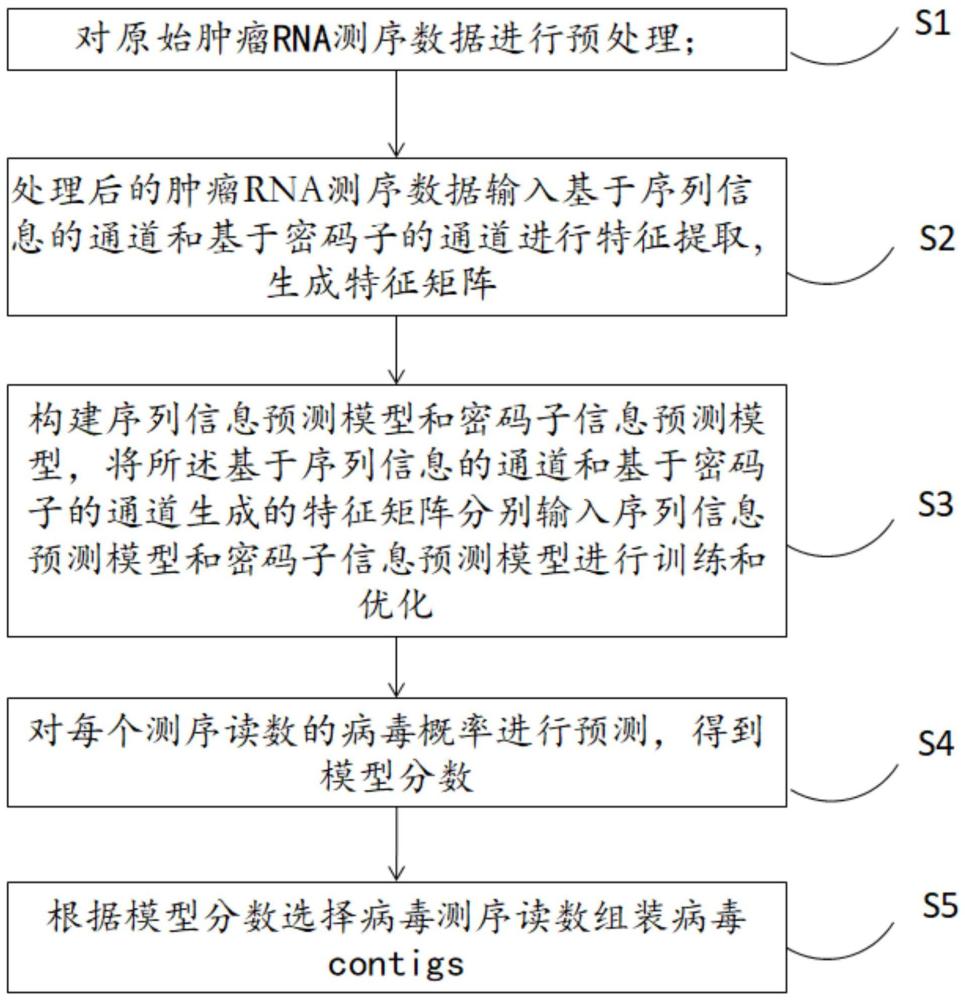
3、s1、对原始肿瘤rna测序数据进行预处理;
4、s2、处理后的肿瘤rna测序数据输入基于序列信息的通道和基于密码子的通道进行特征提取,生成特征矩阵;
5、s3、构建序列信息预测模型和密码子信息预测模型,将所述基于序列信息的通道和基于密码子的通道生成的特征矩阵分别输入序列信息预测模型和密码子信息预测模型进行训练和优化;
6、s4、对每个测序读数的病毒概率进行预测,得到模型分数;
7、s5、根据模型分数选择病毒测序读数组装病毒contigs。
8、本发明通过引入多模态深度学习方法,提高了病毒监测的精度和鲁棒性,同时能够自适应处理不同来源和不同长度的测序数据,有效地应对了现有技术所存在的问题。从而更好地满足医疗和研究领域对于肿瘤测序数据中病毒鉴定的需求。
9、优选的,s1中的预处理包括将所述原始肿瘤rna测序数据人类基因组和phix噬菌体(nc_001422)基因组进行比对匹配,保留没匹配到的测序读数。
10、进一步优选的,在所述基于序列的通道中,使用标记嵌入和位置嵌入生成两个嵌入矩阵并整合为特征矩阵,将序列读数转换为数值表示ms;
11、在所述基于密码子的通道中,构建了一组包含64种不同密码子的滤波器,转换为独热矩阵进行测序读数的特征表示。
12、进一步优选的,所述密码子转换为独热矩阵的过程如下:
13、f(m)=relu(conv2d(f,m)-2)
14、其中cony2d(f,m)被定义为relu表示修正线性单元,i和j表示变量,fn,i,j表示第n个滤波器矩阵第i行第j列元素,mi,j表示独热编码第i行第j列元素。
15、进一步优选的,所述序列信息预测模型包括三个transformer编码块,每个编码块包括多头注意力子层和全连接前馈子层,当输入经过序列信息预测模型时,先经过第一个编码块的多头注意力子层,其输出接着进行残差连接和层标准化后输入第一个编码块的全连接前馈子层,第一个编码块的全连接前馈子层的输出进行残差连接和层标准化输入第二个编码块的多头注意力子层,其输出接着进行残差连接和层标准化后输入第二个编码块的全连接前馈子层,第二个编码块的全连接前馈子层的输出进行残差连接和层标准化输入第三个编码块的多头注意力子层,其输出接着进行残差连接和层标准化后输入第三个编码块的全连接前馈子层,第三个编码块的全连接前馈子层的输出进行残差连接和层标准化得到最终输出;
16、所述密码子信息预测模型输入层为二维卷积层,采用relu激活函数,第二层为flatten层,进行特征打平操作,第三层为最大池化(maxpool)层,第四层为归一化(batchnorm)层,第五层为一维卷积层,采用relu激活函数,第六层为最大池化(maxpool)层,第七层为归一化(batchnorm)层,第八层为一维卷积层,采用relu激活函数,第九层为全局平均池化(adativeavgpool)层,第十层为flatten层。
17、将所述基于序列信息的通道生成的特征矩阵输入序列信息预测模型后,注意力机制基于查询(q)和一组键-值对(k-v)计算注意力分数attension(q,k,v):
18、
19、q=fc(mswq)
20、k=fc(mswk)
21、v=fc(mswv)
22、其中,wq,wk,wv为可学习的参数,dk为k的维度,fc()表示全连接层,softmax()表示将每个元素转为非负值,同时确保所有元素的总和为一。
23、计算多头注意力每个head对应的注意力分数attension(q,k,v),将所有的注意力分数连接在一起,得到多头注意力分数:
24、multihead(q,k,v)=concat(head1,...,headh)wo
25、其中,wo为可学习参数,head1,...,headh表示从第1到第h个head对应的注意力分数attension(q,k,v)。
26、将所述基于密码子的通道转换生成的特征矩阵输入到密码子信息预测模型中用于对读数序列进序列进行评分,得到预测pc。
27、进一步优选的,综合通过序列信息预测模型获得的基于序列信息的预测pt与通过密码子信息预测模型获得的基于密码子的预测pc,通过f(pt,pc)=max(pc,pt)整合两个预测的最大值,得到模型分数。
28、进一步优选的,基于碱基匹配原则,选择模型分数大于0.6的子串拓展种子contigs的左右侧,最后进行拼接,获得完整的病毒contigs。
29、根据本发明的一方面,提出了一种对肿瘤rna测序数据的病毒检测系统,包括,
30、数据预处理模块:对原始肿瘤rna测序数据进行预处理;
31、特征提取模块:将处理后的肿瘤rna测序数据输入基于序列信息的通道和基于密码子的通道进行特征提取,生成特征矩阵;
32、模型构建模块:构建序列信息预测模型和密码子信息预测模型,将所述基于序列信息的通道和基于密码子的通道生成的特征矩阵分别输入序列信息预测模型和密码子信息预测模型进行训练和优化;
33、预测评分模块:对每个测序读数的病毒概率进行预测,得到模型分数。
34、根据本发明的一方面,提出了一种计算机可读介质,其上存储有计算机程序,所述计算机程序在被处理器执行时实施如第一方面中任一实施方式所述的方法。
35、与现有技术相比,本发明的有益成果在于:
36、本发明通过引入多模态深度学习方法,利用两个独立通道处理不同类型的测序数据,使得模型能够同时考虑不同数据来源的信息,提高了病毒监测的精度和鲁棒性,从而提高了识别精度。同时能够自适应处理不同来源和不同长度的测序数据,有效地应对了现有技术所存在的问题。在病毒数据库有限的情况下,本发明能够有效地识别新的潜在病毒,不仅可以处理单细胞rna测序数据,还有潜力处理bu1k rna测序数据,进一步扩大了其应用领域,从而更好地满足医疗和研究领域对于肿瘤测序数据中病毒鉴定的需求。
- 还没有人留言评论。精彩留言会获得点赞!