一种宏基因组测序数据分析方法、计算机介质、系统与流程
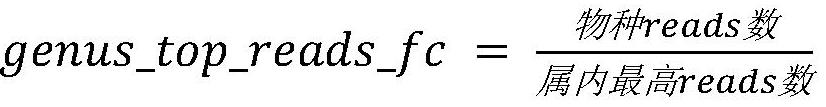
本发明涉及微生物宏基因组测序检测,具体涉及一种宏基因组测序数据分析方法、计算机介质、系统。
背景技术:
1、感染性疾病是临床常见疾病,而病原体诊断是其诊治中的关键环节。传统的病原体鉴定方法包括培养分离、形态学检测、免疫学检测以及核酸检测,是针对一种或几种病原体目标性的鉴别,检测方法覆盖范围小,一次仅能针对一种或几种特定的病原体,且耗时长、阳性率低,难以满足临床需求。随着测序技术的进步和成本的降低,越来越多的微生物得以测序。宏基因组二代测序(metagenomic next-generation sequencing,mngs)作为一种新型病原检测方法,既不依赖于传统的微生物培养,也不需要特异性扩增,且其检测范围广泛,可检测细菌、病毒、真菌、寄生虫、罕见病原体,甚至未知病原体,只要是数据中物种都能覆盖,不需要预先猜测可能的病原体。
2、mngs具有随机、无偏倚特征,可以准确获得检测样本中所有核酸信息,与已知的微生物序列数据库进行比对分析(比对:指将测序的序列与参考基因组进行匹配的过程),根据序列信息鉴定样本中所含的所有病原微生物,分析出致病病原体,指导临床诊断和治疗,预防病情进一步恶化,尤其在疑难、罕见感染性疾病中发挥着重要作用。目前,mngs已被用于呼吸系统、中枢神经系统、血流感染、骨和关节、眼部感染等多系统感染性疾病的诊断和病原鉴定。
3、然而,目前的mngs检测是基于物种比对的方法,为了保证检测范围囊括尽量全面的病原体微生物,各实验室所构建的数据库极为庞大,物种数量往往超过两万种;由于各类微生物的进化关系极为复杂,存在相当多的物种具有相似的序列,会产生大量的物种比对结果,但其中导致患者致病的真实病原很少,其他检出结果往往是背景菌、定植菌或假阳性结果,会导致在物种比对的过程种产生大量的假阳性结果,对真正致病的微生物判断造成干扰。各实验室一般会设定不同的背景菌库、定植菌库以及阴性对照等方法过滤部分结果,但产生假阳性结果的原因很多,无法完全避免假阳性结果的影响,例如:超过2万个物种的庞大数据库中可能存在部分低质量基因组,大量物种因进化关系具有高同源性的基因组,基因组测序组装过程中产生污染和错误,宏基因组二代测序产生的错误,比对软件和算法本身无法做到完全准确,也会产生少量错误。
4、针对假阳性产生的原因,各实验室往往在优化数据库的基础上,通常会通过在宏基因组测序数据处理过程中增加数据过滤和置信度判断的过程,以一些具有统计学意义的指标作为数据过滤和置信度指标,通过设置阈值的方式实现数据过滤,控制假阳性,。一般的阈值选择如鉴定reads数、rpm值、丰度、覆盖度、深度等,因病原种类和基因组大小的差异存在一定的局限性。例如:肺炎克雷伯的基因组大小约为5mb,人疱疹病毒的基因组大小约为150kb,相同拷贝数的情况下,即使能够做到准确鉴定,reads数、rpm等值也会存在较大差异,传统阈值很难在这种情况下准确区分真假阳结果。
5、因此,针对降低宏基因组数据分析假阳性,提出创新的过滤指标,是目前行业内亟待解决的问题之一。
技术实现思路
1、为了实现上述目的,本发明的目的在于提供一种宏基因组测序数据分析方法,reads比对到基因组上的离散程度联合属内最高reads数比值,作为数据过滤条件,设置阈值,降低假阳性。
2、本发明目的之二在于提供一种宏基因组数据分析计算机介质,包括数据过滤模块,运行本发明提供的数据分析方法,降低假阳性。
3、同时,本发明的目的之三在于提供一种宏基因组数据分析计算机系统,包括本发明提供的计算机介质,运行本发明提供的数据分析方法,降低假阳性。
4、为了实现上述目的,本发明采用的技术方案如下:
5、一种宏基因组测序数据分析方法,包括:
6、1)将待分析数据与微生物数据库基因组序列进行比对,计算物种序列离散度和属内最高reads数比值;
7、2)以物种序列离散度和属内最高reads数比值设置筛选条件,符合结果输出条件的,输出物种判定结果。;
8、其中属内最高reads数比值
9、其中物种reads数是鉴定到物种的绝对reads数;属内最高reads数是样本中鉴定到的与所述物种同属的reads数最高物种的绝对reads数。
10、可选的,所述序列离散度计算包括:
11、1)基因组窗口划分:将微生物数据库每个物种基因序列划分为n个窗口;
12、2)各窗口reads分布计算:统计比对到各个窗口的reads数,根据reads的比对结果判断每条reads比对到基因组的窗口位置,计算每个窗口中reads数bin_reads占比对到该基因组的reads总数all_reads的比例,
13、3)离散度计算:统计上述每个窗口的bin_ratio在基因组全部窗口上的分布情况,计算标准差:
14、
15、其中:为所有有reads覆盖窗口的bin_ratio的平均值,n为有reads覆盖窗口的个数,xi为第i个窗口的bin_ratio值。
16、作为优选的,所述基因组窗口划分具体为将微生物数据库每个物种基因序列均匀划分为100个窗口。
17、可选的,所述待分析数据为待分析样本采用二代测序方法测序的下机数据初始过滤后的数据。
18、具体的,所述下机数据初始过滤的具体方法包括去除低质量序列、接头序列和宿主序列。
19、可选的,所述样本为体液样本或组织样本。
20、一种计算机介质,包括:
21、1)序列比对模块:用于将待分析数据与微生物数据库基因组序列进行比对;
22、2)序列离散度计算模块:用于计算物种序列离散度;
23、3)属内最高reads数比值计算模块:用于计算属内最高reads数比值
24、
25、4)结果判断输出模块:用于根据设置的序列离散度和属内最高reads数比值筛选条件,判断输出物种判定结果。
26、可选的,所述序列离散度计算模块包括:窗口划分模块:用于将微生物数据库每个物种基因序列划分为n个窗口;
27、各窗口reads分布计算模块:用于统计比对到各个窗口的reads数,根据reads的比对结果判断每条reads比对到基因组的窗口位置,计算每个窗口中reads数bin_reads占比对到该基因组的reads总数all_reads的比例,
28、离散度计算模块:用于统计上述每个窗口的bin_ratio在基因组全部窗口上的分布情况,计算标准差:
29、
30、其中:为所有有reads覆盖窗口的bin_ratio的平均值,n为有reads覆盖窗口的个数,xi为第i个窗口的bin_ratio值。
31、可选的,还包括数据过滤模块,用于对输入的待分析样本的原始测序数据去除低质量序列、接头序列和宿主序列处理。
32、一种计算机系统,由上述计算机系统组装而成。
33、需要说明的是,上述分析方法、计算机介质和计算机系统,在运行本发明所述离散度计算时,当n=1时,即reads仅比对到一个窗口中,认为离散程度最小,该值赋值为0;
34、本发明宏基因组测序数据分析方法,物种注释分析过程中,以reads比对到基因组上的离散程度(指标1)联合属内最高reads数比值(指标2),作为数据过滤条件,设置阈值,对微生物基因组序列比对结果进行过滤,排除假阳性。
35、本发明设计的过滤指标,充分考虑其生物学意义,理论上,reads覆盖在基因组上的位置离散程度越大,鉴定可信度越高。最理想的情况是鉴定到该病原的大量reads可以均匀分布在基因组的所有位置;若出现大量reads集中比对于一个窗口中,更有可能是reads重复率过高,且比对偏好性造成的影响,可信度低,因此以reads覆盖在基因组上的离散程度作为过滤指标,可以减弱基因组大小差异的影响。同时,考虑物种同源性导致的假阳性结果的影响,相比传统的方法,本发明采用属内最高reads数比值作为过滤指标,评估受属内reads数最高物种同源性带来的干扰,进一步降低假阳性。
36、进一步,本发明基于统计学意义,优化reads覆盖再基因组上的离散程度的计算方法,具体将基因组均匀划分为100个窗口,根据比对结果判断每条reads比对位置位于哪个窗口,通过计算不同窗口比对reads数比例的标准差值来评估reads在基因组上分布的离散程度,该值越小,代表离散程度越高。相比其他数据统计算法,本发明采用标准差值的计算方法,进一步降低假阳性,提高病原体识别准确性。
37、相对传统阈值的过滤效果,本发明两种过滤指标进行结合后可以在保证足够敏感性的同时,有效控制假阳性结果。在模拟数据和临床数据的分析统计中均表现出高于传统方法的性能。
- 还没有人留言评论。精彩留言会获得点赞!