基于域分离网络的lncRNAm6A甲基化位点预测网络模型确定方法及预测方法
本发明属于生物信息,具体涉及一种lncrna m6a甲基化位点预测网络模型确定方法及预测方法。
背景技术:
1、lncrna是一类在基因表达调控、染色质重塑、细胞分化和发育等生物过程中发挥作用的重要分子。lncrna m6a甲基化可以影响肿瘤干细胞的干性维持、肿瘤细胞的侵袭转移、lncrna的稳定性和亚细胞定位等,深入研究lncrna m6a甲基化位点不仅可以揭示lncrna的功能机制,拓展rna表观遗传学研究领域,而且对于疾病诊断与治疗、药物研发等方面的深入理解具有重要意义。
2、近年来,随着生物信息学的不断发展和生物实验数据的不断积累,已经出现了大量用于预测rna m6a甲基化的识别方法。目前已有的rna m6a识别方法大致分为两类:一种是基于机器学习的方法,将rna序列数据经one-hot、理化性质、基因组衍生特征等方法进行编码,使用最小冗余最大相关性(mrmr)、最大关联最大距离(mrmd)等方法进行特征的融合,最后使用支持向量机(svm)、随机森林(rf)等方法进行分类。例如,methyrna模型利用核苷酸的化学性质及其累积的频率信息编码rna序列,并使用svm分类器预测酿酒酵母的甲基化修饰位点。irna-m6a模型通过理化性质矩阵、one-hot编码以及核苷酸化学性质和累计频率编码,采用mrmr算法提取特征并通过svm作为预测分类器的方法,构建了一个在人类、小鼠和大鼠组织有良好泛化能力的预测模型。
3、另一种是基于深度学习的方法,通过模型自行提取rna片段的特征,无需手动设计特征提取器。例如,在im6a-ts-cnn模型中,样本采用one-hot编码方案进行编码,通过一个包含200个过滤器的卷积层和一个大小为4的池化层构成的神经网络构建预测模型,用于预测人,小鼠,大鼠的不同组织的m6a位点。deepm6aseq模型将one-hot编码的序列特征,交由包含两个卷积层和一个bi-lstm网络的深度神经网络在人,小鼠,斑马鱼多个数据集上进行训练和预测。
4、这些方法基本上都是针对泛化rna m6a位点的预测,专门针对lncrna m6a甲基化位点的研究相对较少。lncrna和mrna的m6a在结构、功能、分布等方面存在较大差异,这也就导致使用现有预测模型在提取lncrna m6a的特征时存在挑战,特征提取不完整,最终在预测lncrna m6a时其性能受到限制,影响了lncrna m6a的预测准确性。
技术实现思路
1、本发明的目的是:针对现有rna m6a预测方法缺少专门针对lncrna m6a甲基化研究处理,使得模型对于lncrna m6a甲基化位点的特征提取有所不足,导致位点识别的性能较低的问题。
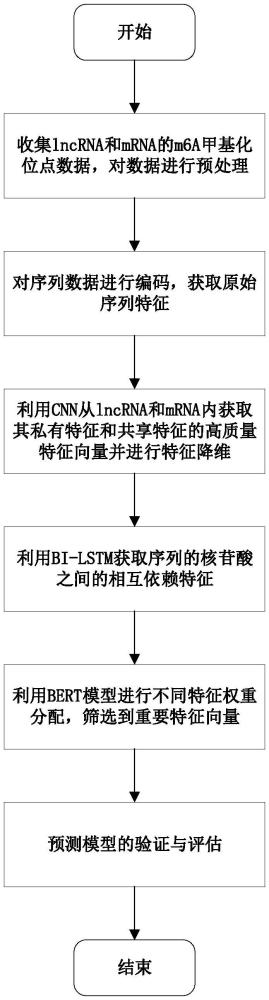
2、基于域分离网络的lncrna m6a甲基化位点预测网络模型确定方法,包括以下步骤:
3、s1、获取lncrna m6a和mrna m6a的位点信息,根据样本位点信息获取对应的rna序列片段,使用cd-hit工具进行去冗余处理,获取到lncrna和mrna的序列片段集合作为样本集,样本集包括阳性样本和阴性样本;以lncrna的阳性样本集和阴性样本集作为目标域的训练数据集,以mrna的阳性样本集和阴性样本集作为源域的训练数据集;
4、s2、针对lncrna,使用one-hot、化学性质与频率信息和位置特异性信息编码方法对序列样本集进行编码,将三种编码方式得到的特征向量对应组合得到lncrna的原始特征向量,记为xt;针对mrna,采用相同的方式得到mrna的原始特征向量,记为xs;
5、s3、创建域分离网络模型,包含目标域lncrna的私有特征提取网络源域mrna的私有特征提取网络目标域lncrna和源域mrna的共享特征提取网络ec;
6、目标域lncrna的私有特征提取网络源域mrna的私有特征提取网络目标域lncrna和源域mrna的共享特征提取网络ec为三个同构的特征提取网络;
7、目标域的私有特征提取网络用于提取lncrna的私有特征
8、源域的私有特征提取网络用于提取mrna的私有特征
9、共享特征提取网络用于提取lncrna和mrna二者的共享特征和
10、s4、将经过目标域lncrna的私有特征提取网络提取到的lncrna的私有特征向量和共享特征提取网络ec提取到的lncrna的共享特征向量组合在一起,经过解码器还原得到还原向量
11、针对mrna的解码采用与lncrna解码进行相同的处理得到mrna的还原向量
12、s5、训练域分离网络模型,每次循环使用lncrna和mrna的数据交替对域分离网络模型进行训练;
13、针对lncrna数据,计算来自lncrna的共享特征向量与私有特征向量的损失域分类的相似性损失lncrna被还原特征向量与原始特征向量xt的损失lncrna的分类损失根据lncrna的总体损失lt进行优化;
14、
15、其中,α,β,γ是控制损失项相互作用的权重;
16、针对源域mrna数据,采用与目标域lncrna损失相同的方式计算总体损失ls进行优化;
17、域分离网络模型训练完成后,提取训练好的域分离网络模型中的共享特征提取网络ec再连接一个分类器,得到用于lncrna m6a甲基化位点预测的网络模型。
18、进一步地,s1所述获取到lncrna和mrna的序列片段集合作为样本集的过程包括以下步骤:
19、s11、获取到lncrna和mrna的m6a的位点信息;
20、s12、根据数据库提取到的数据文件所包含的染色体号、起始位置、终止位置、正负链标识表示,从hg38人类基因组中提取到多条rna序列片段,然后截取序列片段,使用cd-hit工具剔除与其他序列具有高相似性的序列片段,所述具有高相似性是指相似性超过相似性阈值的序列片段,将最终得到的序列片段集合作为模型训练的阳性样本集;
21、s13、在阳性样本位点的上下游截取以腺嘌呤为中心的序列片段,然后使用cd-hit工具剔除与其他序列具有高相似性的序列片段,以及与阳性样本位点重叠的片段,将最终得到的序列片段集合作为模型训练的阴性样本集。
22、进一步地,使用one-hot编码方法对序列样本集进行编码的过程中,将acgu四种核苷酸表示为四维二进制向量;然后将长l的rna序列中每个核苷酸编码并串联起来,得到4*l维的特征向量表示xone-hot。
23、进一步地,使用化学性质与频率信息编码方法对序列样本集进行编码的过程包括以下步骤:
24、使用三个坐标(x,y,z)来表示四种核苷酸的化学性质,x坐标表示环结构,y坐标表示官能团,z坐标表示氢键,并为这三个坐标赋值0和1;rna序列中的第i个位置核苷酸s用s=(xi,yi,zi)编码;
25、然后进行核苷酸积累频率编码,所述核苷酸积累频率被定义为第i个位置的核苷酸在第i个位置之前出现的频率;第i个位置的核苷酸的积累频率fi=di/i,其中di为第i个核苷酸在前i个核苷酸中出现的次数之和;
26、将长l的rna序列中每个核苷酸依据化学性质和核苷酸累计出现频率进行编码并组合起来,每个rna序列得到4*l维的特征向量表示xncpf。
27、进一步地,使用位置特异性信息编码方法对序列样本集进行编码的过程包括以下步骤:
28、位置特异性倾向用来描述k-mer核苷酸在特定位置上出现的倾向性或偏好性;
29、在k-mer取值1时全局特征矩阵的第i列可计算为阳性和阴性样本中第i位4种核苷酸的出现频率之差,如下式所示:
30、
31、其中,i表示rna片段的第i个序列位置,f+(1:4,i)和f-(1:4,i)分别表示阳性和阴性样本中4种核苷酸(a,u,c,g)在第i个位置出现的频率,分别表示阳性数据集中核苷酸(a,u,c,g)在第i个序列位置出现的频率,分别表示阴性数据集中核苷酸(a,u,c,g)在第i个序列位置出现的频率;
32、rna片段长度为l,基于fpsnp(1:4,i)得到一个4*l的全局特征矩阵;然后根据序列位置对应核苷酸在全局特征矩阵中的值来编码rna片段,得到一个l维的位置特异性倾向特征;
33、在k-mer取值2时得到rna序列的位置特异性倾向全局特征矩阵,如下式所示:
34、
35、其中,j表示rna片段的第j个序列位置,f+(1:16,j)和f-(1:16,j)分别表示阳性和阴性样本中16种二核苷酸(aa,au,ac,…,gg)在第j个位置出现的频率,分别表示阳性数据集中二核苷酸(aa,au,ac,…,gg)在第j个序列位置出现的频率,分别表示阴性数据集中核苷酸(aa,au,ac,…,gg)在第j个序列位置出现的频率;
36、rna片段长度为l,基于fpsdp(1:16,j)得到一个16*(l-1)的全局特征矩阵;然后,根据序列位置对应核苷酸在全局特征矩阵中的值来编码rna片段,得到一个l-1维的位置特异性倾向特征;
37、以相同的方式,在k-mer取值3时得到长度为l的rna序列的位置特异性倾向全局特征矩阵,其维度为64*(l-2);根据序列位置对应核苷酸在全局特征矩阵中的值来编码rna片段,得到一个l-2维的位置特异性倾向特征;
38、最后将k-mer取值1、2、3时得到的序列的特征串联在一起得到序列的位置特异性倾向特征表示xpsp。
39、进一步地,计算来自lncrna的共享特征向量与私有特征向量的损失域分类的相似性损失lncrna被还原特征向量与原始特征向量xt的损失lncrna的分类损失的过程包括:
40、s51、计算
41、
42、其中是frobenius范数的平方,矩阵为lncrna样本中的共享特征向量,即矩阵为lncrna样本中的私有特征向量,即
43、s52、将lncrna的共享特征进行梯度反转,然后通过全连接网络完成域分类将目标域的域分类定义为1;计算
44、
45、其中,nt表示目标域lncrna的样本数量;
46、s53、计算
47、
48、s54、计算
49、
50、其中,是样本输入z的类标签的独热编码,模型的预测标签。
51、进一步地,所述三个同构的特征提取网络均包括cnn网络层、bi-lstm网络层和bert网络层。
52、进一步地,s4所述的解码器包括两个卷积层。
53、进一步地,用于lncrna m6a甲基化位点预测的网络模型中的分类器为训练过程中共享网络对应的用于预测分类结果的分类器。
54、基于域分离网络的lncrna m6a甲基化位点预测方法,针对待预测的lncrna首先得到对应的原始特征向量xt,然后利用所述的基于域分离网络的lncrna m6a甲基化位点预测网络模型确定方法得到用于lncrna m6a甲基化位点预测的网络模型,并基于用于lncrnam6a甲基化位点预测的网络模型实现lncrna m6a的预测。
55、本发明的有益效果是:
56、本发明利用mrna m6a有丰富先验信息的优势特征,经迁移学习辅助预测lncrnam6a甲基化位点,提升了模型的预测性能。本发明提取人类的lncrna和mrna的m6a甲基化位点信息,经过one-hot、化学性质与频率、位置特异性倾向编码,提取了序列的不同角度的特征信息,借助cnn、bi-lstm、bert网络层构成的特征提取模块,实现了多元特征的有效融合,有效获取了甲基化修饰的有效特征。域分离网络获取了来自lncrna的私有特征,mrna的私有特征以及lncrna和mrna的共享特征信息,通过控制损失信息,使得共享模块尽可能提取到来自lncrna和mrna的共享特征表示。在基准数据集以及不同物种数据集上的验证结果表明,借助mrna丰富先验信息优势进行辅助预测,模型对于lncrna m6a甲基化位点的预测性能得到了提升,具有较高的泛化能力和鲁棒性。
- 还没有人留言评论。精彩留言会获得点赞!