基于同源性分析和区域加权的线粒体基因组重排量化方法
本发明属于分子生物学和生物信息学,更具体地说,涉及一种基于同源性分析和区域加权的线粒体基因组重排量化方法。
背景技术:
1、对线粒体基因组结构特征进行量化分析,对于我们深入理解其复杂性至关重要。准确量化和阐释组织多样性,有助于我们发现生物演化规律和模式。当前,较为通用的量化线粒体基因组重排的方法为qmgr(quantifying mitochondrial genome rearrangement)。该方法将单个基因视为一个结构单位,通过计算其相邻的两个基因的变化,全局性地等价量化分类单元中的多个重排特征。除了对特定分类群的重排量化分数(rs)进行计分外,该方法还能够计算线粒体基因组中单个基因的重排频率(rf),以便我们检测分类单元内的保守基因簇和高频重排基因簇。但是由于其计分策略是基于相邻两个基因的变化而制定的,因此其计分结果会受到相邻两个基因状态的影响,这可能导致分数出现偏高或偏低的情况。具体来说,计分偏差情况可以分为以下四种:(1)对于某个基因,如果其本身没有发生变化,但是其相邻的两个基因发生了变化,会导致该基因分值偏高。(2)在遍历每个待量化基因序列时,该方法会对基准序列中相应的每一个基因进行检索,并把分值添加到基准基因中,如果基准序列中不存在某基因,则待量化基因序列中无法检索到该基因,这将会导致基准序列分值偏高。(3)如果基因簇发生整体位移,则基因簇内的基因分数偏低。(4)该方法无法对发生长距离位移的基因赋予相应的权重,从而导致分值偏低。这四种情况共同影响现有方法qmgr结果的准确性,也将导致我们对保守基因簇和高频重排基因簇的判断出现偏差。
2、公告号为cn117133351b专利文献公开了一种优化的线粒体基因重排量化方法,其主要是采用划分数组将基因序列分段并使用滑动窗口来计算每个基因的得分,允许对重排强度各异的基因簇给予不同权重,但其只解决了现有方法qmgr的第四种情况的问题,前三种情况中由于计分策略导致的结果存在误差的问题仍未解决,并且其不是基于基因序列的同源性进行量化,不能准确地描述基因组结构的多样性。
技术实现思路
1、针对上述现有技术中存在的不足,本发明提供一种基于同源性分析和区域加权的线粒体基因组重排量化方法,命名为mitoq,旨在量化动物线粒体基因组中基因组组织的多样性。该方法基于基因顺序的同源性,对基因簇进行分区并为跨区基因分配不同的权重,从而能够更准确地描述基因组结构的多样性特征。在对脊椎动物线粒体基因组数据测试中,本发明方法mitoq表现出优于现有方法qmgr的准确性和可解释性。这一方法不仅显著提升了基因组的可比较性,能更准确地揭示出不同类群线粒体基因组结构的差异性,同时在发现基因组的演化规律、探索基因组功能及研究分子演化过程等方面,也具有重要的科学价值。更为关键的是,本发明方法mitoq为基因组学研究和生物演化研究开辟了一个有别于传统序列演化的全新视角,为相关领域的研究提供了新的思路和方法。
2、为实现上述目的,本发明采用的技术方案如下。
3、一种基于同源性分析和区域加权的线粒体基因组重排量化方法,包括以下步骤:
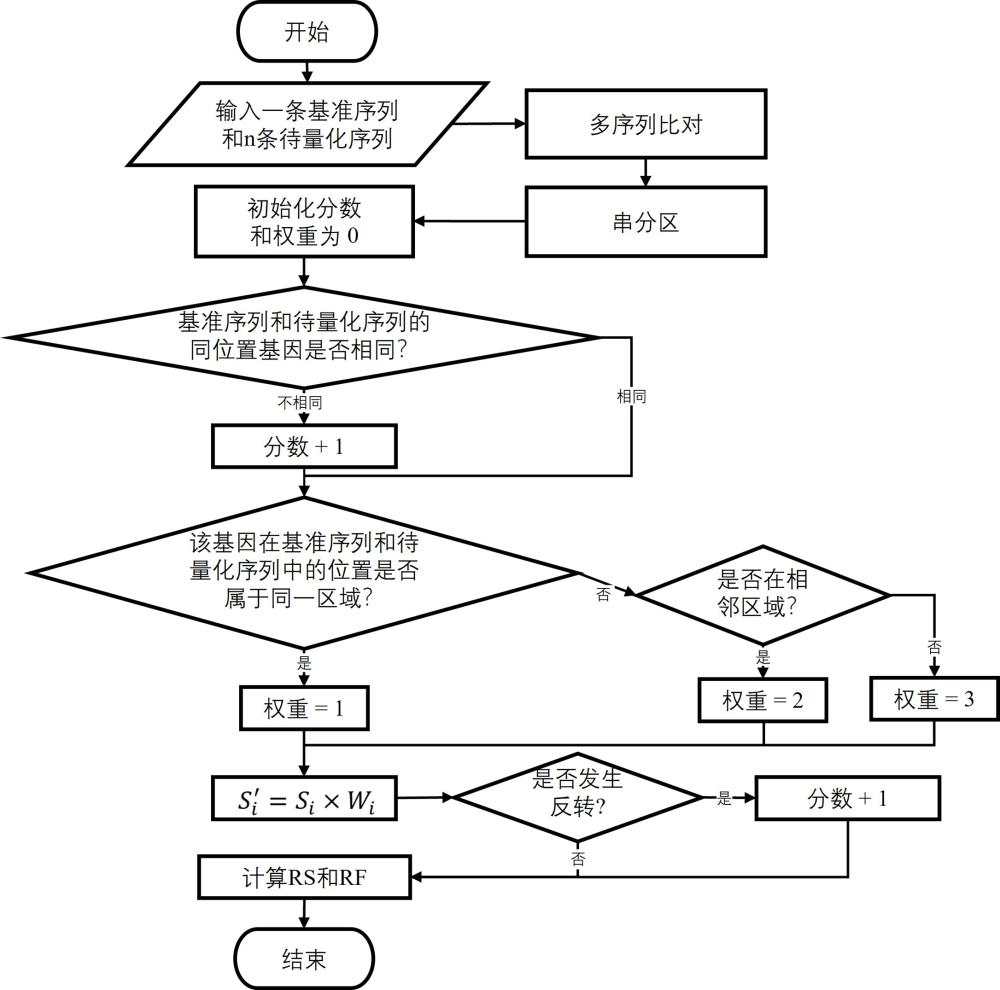
4、步骤s1,输入一条基准序列和n条待量化序列;
5、步骤s2,手工多序列比对,将基准序列与n条待量化序列对齐;
6、步骤s3,对照基准序列,将n条待量化序列从控制区到复制起始点进行分区;
7、步骤s4,设待量化序列中第i个位置上基因的重排分数的初始值为0,权重的初始值为0,将待量化序列和基准序列的同位置基因进行比较,如果相等,则将待量化序列中第i个位置上基因的重排分数计为0分;如果不相等则计为1分,得到待量化序列中第i个位置上基因的初步重排分数;然后作以下两个判断:
8、步骤s41,对待量化序列中第i个位置上的基因在待量化序列和基准序列中的区域进行判断,赋予权重,若在同一区域给予权重的值为m1,若在相邻区域给予权重的值为m2,若跨区域则给予权重的值为m3,且m1<m2<m3,将权重与已计算得到的待量化序列中第i个位置上基因的初步重排分数相乘得到重排分数;
9、步骤s42,判断待量化序列中第i个位置上的基因是否发生了反转,如果发生了反转就给重排分数加1分,否则保持不变,得到待量化序列中第i个位置上基因的最终重排分数;
10、步骤s5,按照步骤s4~步骤s42计算得到每条待量化序列中每个基因的最终重排分数,最后将每条待量化序列中所有基因的最终重排分数相加得到每条待量化序列的重排量化分数rs,将所有待量化序列中每种相同基因的分数相加得到每种基因的单基因重排频率rf。
11、其中,所述步骤s2中,手工多序列比对,将基准序列与n条待量化序列对齐的具体方法为:通过填补空位的方式将基准序列与n条待量化序列的基因串中缺失的基因的数量补齐,将基因在垂直方向上对齐,使基因组中所有基因串的长度都与最长的序列中基因串的长度一致。通过手工对齐基准序列和待量化序列,确保其正确比对,为后续分析提供准确的基础。
12、其中,所述步骤s3中,根据控制区和复制起始点进行分区的分区方式是出于生物学方面的考虑并经过多次实验分析后采用的分区策略。划分出的区域数量大于等于2。
13、其中,所述步骤s4中,通过对每个基因位置进行比较,并考虑区域位置,对基因的位移情况精确量化,能确保对短距离位移和长距离位移的基因进行区分从而进一步给予相应的权重。
14、其中,所述步骤s42中,引入反转判断机制,判断基因的反转情况,反转在基因的重排中较为常见,因而对反转这一事件同样给予分数,能够进一步精确量化基因重排的程度。
15、其中,待量化序列中第i个位置上基因的最终重排分数的计算公式如下:
16、;
17、其中,为待量化序列中第i个位置上基因的初步重排分数,为待量化序列中第i个位置上基因的权重;
18、待量化序列中第i个位置上基因的权重的赋予规则的公式表示如下:
19、;
20、待量化序列中第i个位置上基因的初步重排分数的计分规则的公式表示如下:
21、。
22、本发明方法mitoq解决了现有方法qmgr中存在的四种主要问题,并已被应用于分析以往线粒体基因组研究中的一个综合数据集。通过成对基因顺序比较和区域加权技术,本发明方法mitoq精准地捕捉了基因组组织的变化动态。为了进一步验证其性能优势,我们将本发明方法mitoq所产出的结果与现有方法qmgr的结果进行了详细对比,并采用图表直观展示了这两种方法在同一数据集上的应用效果。通过对比我们发现,由于现有方法qmgr评分规则的局限性,所有分类群均普遍存在的相邻基因影响计分分值的情况。特别是在鸟类数据分析中,单个基因除了受相邻基因影响外,基准序列中不存在的基因也被计入较高分值以及基因簇发生整体位移导致簇内基因分数偏低的情况也较为严重,这显著导致了结果的大幅偏差。具体而言,由于现有方法qmgr对缺失基因的评分机制存在缺陷,使得得分虚高,因而未能对基因簇的整体位移进行正确计分,使得簇内基因得分低于预期。此外,除鸟类外的其他四个类群均无法对发生长距离位移的基因进行精准计分,在不同程度上导致了评分过低。本发明方法mitoq则不存在上述问题。
23、本发明的有益效果:
24、(1)本发明方法mitoq对线粒体基因组输入数据的约束条件较少,因此能够准确识别并统计真实数据中的几乎所有情况,包括但不限于基因重排、基因复制或丢失并允许少量基因含量的变化;确保了数据分析的全面性和准确性。
25、(2)本发明方法mitoq是一种基于基因顺序的同源性量化方法,其量化结果能更好地推断出物种的同源性关系,为生物学研究提供了更可靠的科学依据。
26、(3)本发明方法mitoq引入了区域加权的思想,能够对发生长距离位移的基因赋予不同的权重,这一设计不仅使结果更符合生物学原理,还能提高该方法对基因组动态的敏感度和解释力。
27、本发明方法解决了现有方法qmgr中存在的全部问题并基于基因序列的同源性进行量化,能够更准确地描述基因组结构的多样性。这一方法不仅增强了基因组的可比较性,可以更准确地展示出不同类群线粒体基因组结构的差异性,在揭示基因组的演化规律、探索基因组功能、研究分子演化过程等方面具有重要的科学价值,而且为基因组学研究、生物演化研究提供了有别于序列演化的视角,为相关领域的研究提供新的思路和方法。例如,本发明的基因组重排量化方法可以应用在以下几个主要领域:
28、(1)遗传性疾病研究领域:某些遗传性疾病与基因组结构变异有关,应用重排量化方法可以发现这些异常结构变化,有助于疾病机理的理解。
29、(2)进化基因组学领域:比较不同物种或群体间基因组结构的差异,可以推测物种或群体的进化历程和关系。
30、(3)基因组工程领域:合成生物学领域利用基因组重排技术进行基因组改造,为生物制药、生物能源等应用提供新的可能。
31、(4)人群遗传学研究领域:大规模人群基因组结构变异分析有助于探讨人类遗传多样性和族群差异。
32、(5)农业基因组学领域:研究作物和家畜基因组结构变异,可以为品种改良和遗传育种提供依据。
- 还没有人留言评论。精彩留言会获得点赞!