基于迁移学习和三通道组合GNN预测蛋白质功能的方法
本发明涉及深度学习和生物信息学领域,具体涉及一种基于迁移学习和三通道组合gnn预测蛋白质功能的方法。
背景技术:
1、蛋白质是生命活动与细胞代谢的必需品,对蛋白质功能的研究将直接阐明蛋白质在生理或病理中的各种特殊机制,揭示蛋白质的合成、降解、加工、修饰的过程,使得人们能够更好的理解基因的表达与调控。目前主要通过酶活性测定、细胞生物学实验等技术测定蛋白质功能,基于基因本体论(gene ontology, go)将蛋白质功能划分为:分子功能(molecular function, mf);生物过程(biological process, bp);细胞组分(cellularcomponent, cc)三个大类。
2、随着x射线单晶衍射、冷冻电子显微镜等蛋白质测序及其结构解析技术的发展,以及蛋白质结构预测模型的应用,已知的氨基酸序列和结构数量与相应的功能注释相差巨大,为了避免直接通过实验测定蛋白质功能产生的高昂设备和时间成本,研究人员越来越多地转向使用计算方法进行功能预测。蛋白质的序列和分子结构特征在一定程度上决定了其拥有的特定功能,通过结合氨基酸序列或分子结构信息,设计高效且精准的蛋白质功能预测方法成为近年来对蛋白质功能研究的重点。
3、一开始研究者通过比对序列的相似性和同源性来推测蛋白质的功能,即拥有高同源性的序列之间,它们的功能也大概率相近,可将它们已知功能注释转移到相似蛋白。基于此理论,有研究人员开发出blast2go工具利用序列相似性进行功能注释转移和分析。也有方法通过比对结构的相似性来推测蛋白质拥有相似的功能,有文献提出通过接触图计算蛋白质结构之间的相似性从而预测蛋白质功能。
4、根据序列或结构的相似和同源性直接预测蛋白质功能极易出错。随着深度学习技术在多领域问题求解的出色表现,人们开始探索其在蛋白质功能预测中的应用。例如,deepgo模型使用cnn提取氨基酸序列特征,并与string数据库中提取的蛋白质互作用网络结合,预测蛋白质功能。deepgo模型仅针对序列进行特征建模,忽略了蛋白质的分子结构对其拥有的特定功能产生的巨大影响。基于此,有研究者提出了一种结合分子结构预测功能的模型deepfri,从pfam数据库的整个序列集合中采样10m的序列,训练了一个基于lstm的蛋白质语言模型作为氨基酸序列的特征提取器。结合氨基酸序列的残基级特征矩阵和从蛋白质空间结构文件中提取的残基邻接矩阵,输入gcn图卷积神经网络中预测蛋白质功能。然而,lstm在处理一些长序列的文本数据时缺陷明显,且仅使用gcn处理一些复杂的无规则图数据时存在局限性。在实验数据选取上,deepfri仅使用pdb蛋白质数据库中的高精度蛋白质结构,以及swiss - model构建的基于同源性的结构模型,但大多数蛋白质都缺少这些数据。同时在实际操作的时候,由于每一个蛋白质数据库并不直接相关,蛋白质的序列、分子结构、功能注释等关键信息须通过数据库交叉引用从不同的源数据库检索,这极大地阻碍了研究者们完整且便捷的获得蛋白质的关联信息,且一些提供下载的数据库如蛋白质-配体互作用数据库chembl是mysql类型的关系型数据库,在访问便捷性和检索效率上相比文档类型的nosql数据库较低。
技术实现思路
1、针对上述问题,本发明提出了一种基于迁移学习和三通道组合gnn预测蛋白质功能的方法,该方法提高预测精度,同时本发明还提供了一种蛋白质文档型关联数据库,便于高效的获取相关蛋白质在侧重不同的蛋白质数据库中的详细关联信息,基于此用户可以从蛋白质文档型关联数据库中提取由数据驱动的蛋白质下游预测任务的基准数据集。
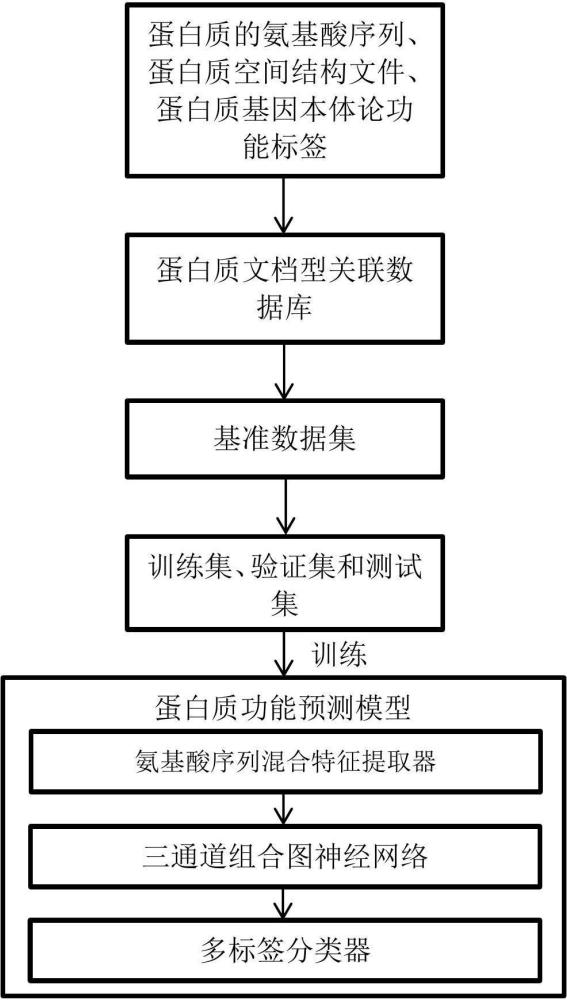
2、本发明所采用的技术方案是:基于迁移学习和三通道组合gnn预测蛋白质功能的方法,包括以下步骤:
3、步骤s1、从蛋白质公共数据库获取蛋白质的氨基酸序列、蛋白质空间结构文件、蛋白质基因本体论(go)功能标签,对所获取的数据进行交叉关联和数据清洗后,构建nosql类型的蛋白质文档型关联数据库,从蛋白质文档型关联数据库中获取蛋白质功能预测任务的基准数据集,并将其划分为训练集、验证集和测试集;
4、步骤s2、解析蛋白质空间结构文件,从中提取蛋白质分子中每一个氨基酸残基中ca原子的空间坐标,基于所有残基中ca原子的空间接触关系,分别按照不同的接触阈值提取出有不同连接密度的氨基酸残基接触图,以映射蛋白质分子的复杂空间结构;
5、步骤s3、基于迁移学习构建氨基酸序列混合特征提取器,通过氨基酸序列混合特征提取器从氨基酸序列中提取出多类残基级特征,组成序列初级嵌入表示;对序列初级嵌入表示进行特征融合和富集化操作后得到氨基酸序列的残基级混合特征映射;
6、步骤s4、引入图卷积网络和拥有多头图注意力机制的gatv2网络构建三通道组合图神经网络,以氨基酸序列的残基级混合特征映射作为三通道组合图神经网络的初始特征矩阵输入,氨基酸残基接触图将作为不同特征通道的邻接矩阵输入,捕捉和学习蛋白质分子中氨基酸残基间的复杂图关系,结合氨基酸序列的残基级混合特征映射和多级结构特征进行蛋白质功能预测;
7、步骤s5、构建一个多标签分类器,对三通道组合图神经网络的输出节点特征进行全局池化以及聚合操作,将聚合后特征映射到0-1的概率空间中,得到每一个蛋白质基因本体论功能标签的概率值;
8、步骤s6、由序列混合特征提取器、三通道组合图神经网络、多标签分类器组成一个完整的蛋白质功能预测模型,在训练集上预训练该蛋白质功能预测模型,使用验证集和测试集评估蛋白质功能预测模型不同训练阶段的性能,使用训练合格的蛋白质功能预测模型进行蛋白质功能预测。
9、进一步优选,步骤s2中所述残基中ca原子的空间接触关系是:一个蛋白质由n个氨基酸残基构成,遍历蛋白质空间结构文件,从中提取出每个氨基酸残基中ca原子的空间结构坐标,基于此坐标计算所有ca原子之间的空间距离,设定距离阈值判定两个残基之间是否接触,由此得到一个n*n的氨基酸残基接触矩阵,随后,只取有接触关系的边r,按照边r的源节点和目标节点转换距离矩阵为2*2r的邻接矩阵,以此作为氨基酸残基接触图。
10、进一步优选,步骤s2中所述不同连接密度的氨基酸残基接触图是:分别计算pdb真实测定的蛋白质空间结构文件和alphafold预测的蛋白质空间结构文件的氨基酸残基接触图,并且根据不同的距离阈值大小计算代表不同接触密度的氨基酸残基接触图。
11、进一步优选,步骤s3中,将蛋白质的完整序列或某一段子序列输入氨基酸序列混合特征提取器中,先由bi-lstm网络提取出的序列嵌入表示,随后,由33层transformer编码器组成的esm2网络继续提取出序列嵌入表示,将bi-lstm网络和esm2网络提取的序列嵌入表示共同作为氨基酸序列的序列初级嵌入表示。
12、进一步优选,步骤s3中所述对序列初级嵌入表示进行特征融合和富集化操作是:对序列初级嵌入表示按类别切片分为:序列独热编码映射、残基序列深度上下文特征和残基生化特性编码,对切片进行可学习的非线性映射,通过调整各自隐藏层的维度大小,对序列初级嵌入表示做特征富集化与降维,最后,通过relu非线性映射以及特征融合操作,得到氨基酸序列的残基级混合特征映射。
13、进一步优选,所述三通道组合图神经网络,由串联两层gcn网络构成第一个特征通道;另一个相同的串联两层gcn网络构成第二个特征通道,第一个特征通道与第二个特征通道平行,构成一个2*2并行双通道gcn网络;引入有多头图注意力机制的gatv2网络组成第三个特征通道,将gatv2网络串联至并行双通道gcn网络之后。
14、进一步优选,所述三通道组合图神经网络的特征聚合和传播方式为:使用氨基酸序列的残基级混合特征映射作为2*2并行双通道gcn网络的初始特征矩阵输入,以更小的接触阈值提取的稀疏氨基酸残基接触图作为该并行网络其中一条特征通道的邻接矩阵输入,以更大的接触阈值提取的密集氨基酸残基接触图作为另外一条特征通道的邻接矩阵输入;将2*2并行双通道gcn网络的输出特征进行特征融合后得到融合特征,将其作为gatv2网络的特征矩阵输入,使用稀疏氨基酸残基接触图作为gatv2网络的邻接矩阵输入;gatv2网络在对融合特征做进一步噪声过滤和特征提取后得到三通道组合图神经网络的输出节点特征。
15、进一步优选,步骤s1中,基于mongodb构建nosql类型的蛋白质文档型关联数据库。
16、进一步优选,构建nosql类型的蛋白质文档型关联数据库的步骤如下:
17、步骤s101、使用接口测试脚本和测试工具postman测试uniprot数据库的开放数据接口,经过筛选条件得到对应蛋白质条目的uniprot数据库标识符:upid,筛选出的蛋白质条目需拥有实验测定的蛋白质空间结构文件或alphafold模型预测的蛋白质空间结构文件,选择蛋白质基因本体论作为蛋白质的功能标签,筛选出来的每条蛋白质条目拥有的蛋白质基因本体论标签总数应大于等于3,筛选出的uniprot数据库标识符作为数据库的源数据id;
18、步骤s102、通过筛选得到的uniprot数据库标识符访问uniprot数据库数据接口,从接口返回的数据包中获取对应蛋白质条目在pdb、alphafold数据库中的交叉索引id,同时通过uniprot数据库标识符关联chembl数据库的目标靶点蛋白信息表的主键靶点id,并以此关联获取chembl数据库中的其余次表id;
19、步骤s103、分类请求上述各类id对应的数据接口,通过beautifulsoup、 jsonpath框架和正则表达式解析接口返回的xml和json数据包,得到uniprot、 pdb、alphafold三个数据库中的蛋白质关联信息组成的json字典以及chembl数据库中的靶点蛋白关联数据组成的json字典;
20、步骤s104、基于json字典,以uniprot数据库标识符作为主键使用pymongo框架搭建蛋白质信息主表,以主键靶点id作为主键搭建包含靶点信息的chembl关联信息表;
21、步骤s105、以chembl关联信息表中的主键靶点id与蛋白质信息主表中的uniprot数据库标识符进行交叉关联,得到最终的蛋白质文档型关联数据库,用户可以仅通过uniprot数据库标识符检索相关蛋白质条目在uniprot、pdb、 alphafold、chembl数据库中的关键信息。
22、本发明的有益效果如下:
23、(1)提出了一种基于mongodb构建nosql类型的蛋白质文档型关联数据库的方法,通过nosql类型的蛋白质文档型关联数据库,用户可以仅根据一个主键靶点id检索相关蛋白质条目的详细关联数据,无需再去到多个数据库中进行交叉索引,极大的提升了蛋白质关联信息的检索效率。且相较于直接下载的完整mysql类型的公共数据库,nosql类型的数据库在访问便捷性上有巨大提升。
24、(2)当下大多数的蛋白质功能预测模型包括deepfri仅使用pdb蛋白质数据库中的高精度蛋白质结构,以及swiss - model构建的基于同源性的结构模型,但大多数蛋白质都缺少相对应的数据,为解决这个问题,从文档型nosql蛋白质关联数据库中获取的实验数据集,可同时拥有由实验测定的pdb高精度结构数据以及alphafold模型预测的结构数据,在降低数据获取成本的同时也可以提升模型的泛化性能。
25、(3)直接通过序列独热编码映射或lstm模型提取的氨基酸序列特征缺乏残基深度上下文信息,本发明基于迁移学习构建氨基酸序列混合特征提取器,迁移学习基于bi-lstm和transformer的两类蛋白质语言模型,结合序列独热编码映射、残基序列深度上下文特征和残基生化特性编码,共同构成对氨基酸序列的序列初级嵌入表示,在模型训练过程中对序列初级嵌入表示进行可学习的非线性映射,缩小实验开销的同时进一步富集化残基特征,由此得到的残基级混合特征映射的特征表达能力将更加强大。
26、(4)deepfri模型架构由gcn网络串联构成,在处理复杂的无规则图如蛋白质空间结构时存在一定局限性,本发明引入有多头注意力机制的gatv2网络,允许蛋白质功能预测模型同时学习多组注意力权重,每个注意力头都可以学习到不同的注意力权重,增强了蛋白质功能预测模型对复杂图结构的表达能力。
27、(5)单特征通道无法高效学习蛋白质分子的多组结构特性,本发明结合gcn网络和有多头图注意力机制的gatv2网络共同构成一个三通道组合图神经网络架构,通过一个2*2的并联gcn网络同时学习两类不同接触阈值提取的残基接触图,随后通过串联的gatv2网络进一步对双通道并联网络输出的特征进行噪声过滤,该架构能够组合学习蛋白质分子的深层次序列和结构特性,更好的捕捉蛋白质结构中残基之间的复杂关系。同时,在模型最后的多标签分类阶段,本发明通过融合两类全局池化信息,使蛋白质功能预测模型能进一步获取更多的节点特征。
- 还没有人留言评论。精彩留言会获得点赞!