基因组拷贝数变异分析数据的处理方法、装置及设备与流程
本发明涉及生物信息,具体涉及一种基因组拷贝数变异分析数据的处理方法、装置及设备。
背景技术:
1、基因组拷贝数变异(copy number variation, cnv)是一种用于识别个体基因组dna序列中相对于参考序列增加或减少的部分的遗传学检测。这些变化可能包括基因的重复、缺失或插入,它们可能与某些遗传性疾病或癌症有关。目前对cnv进行检测的方法主要有基于芯片的比较基因组杂交(array-based comparative genomic hybridization,acgh)、单核苷酸多态性微阵列芯片(single nucleotide polymorphism array, snparray)、多重连接依赖探针扩增(multiplex ligation-dependent probe amplication,mlpa)等,其中mlpa方法是检测染色体拷贝数变异的金标准。随着人类基因组计划的完成,dna测序技术进入高速发展的阶段,基于下一代测序(next-generation sequencing, ngs)技术和生物信息学分析的基因组拷贝数变异检测(copy number variation, cnv)为产前诊断以及遗传病筛查提供了新的手段。
2、cnv是人类基因多样性的重要组成部分,在诊断临床症状不明的患者和鉴定已建立的胎儿染色体疾病综合征方面具有重要作用。高深度的双端全基因组测序(>30×)对cnv检测具有高敏感度及分辨率,但是花费高且试验周期比较长,同时其对计算机的硬性条件及分析人员的要求也比较高。近年的研究发现,利用低深度的全基因组测序技术(1×-5×)甚至是超低深度的全基因组测序技术(0.1×)可对23对染色体的非整倍体变异、大于50kb/100kb/500kb的微缺失/微重复进行检测,用于排查反复自然流产、先天畸形、发育迟缓等多种疾病的遗传病因。相对于高深度测序来说,低深度全基因组测序的成本更低,可以降低检测成本,检测时间短可以快速得到基因组信息。但是,低深度的全基因组cnv分析结果的解读难度大。
技术实现思路
1、有鉴于此,本发明提供了一种基因组拷贝数变异分析数据的处理方法、装置及设备,以解决低深度、甚至超低深度的全基因组cnv分析结果解读难度大的问题。
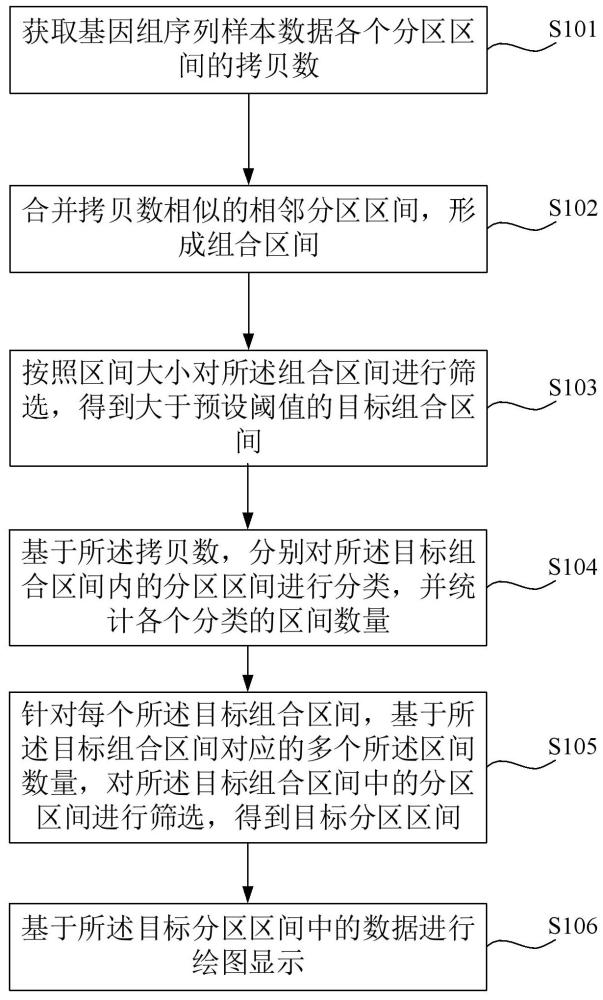
2、第一方面,本发明提供了一种基于超低深度全基因组高通量测序的基因组拷贝数变异分析数据的处理方法,所述方法包括:
3、获取基因组序列样本数据各个分区区间的拷贝数;
4、合并拷贝数相似的相邻分区区间,形成组合区间;
5、按照区间大小对所述组合区间进行筛选,得到大于预设阈值的目标组合区间;
6、基于所述拷贝数,分别对所述目标组合区间内的分区区间进行分类,并统计各个分类的区间数量;
7、针对每个所述目标组合区间,基于所述目标组合区间对应的多个所述区间数量,对所述目标组合区间中的分区区间进行筛选,得到目标分区区间;
8、基于所述目标分区区间中的数据进行绘图显示。
9、在一种可选的实施方式中,所述合并拷贝数相似的相邻分区区间,形成组合区间之前,还包括:
10、对于所述分区区间中的y染色体对应的第一分区区间,若拷贝数小于第一预设阈值,则删除所述第一分区区间。
11、在一种可选的实施方式中,所述第一预设阈值的取值范围为0.4-0.6。
12、在一种可选的实施方式中,所述针对每个所述目标组合区间,基于所述目标组合区间的多个所述区间数量,对所述目标组合区间中的分区区间进行筛选,得到目标分区区间,包括:
13、基于所述目标组合区间的多个所述区间数量,确定对应的分区区间筛选规则;
14、按照确定的所述分区区间筛选规则,对所述目标组合区间中的分区区间进行筛选。
15、在一种可选的实施方式中,对所述目标组合区间中的分区区间进行筛选的分区区间筛选规则为:将所述目标组合区间中拷贝数在预设范围内的分区区间筛选出来,其中,所述预设范围为设定值或为基于所述目标组合区间的拷贝数确定。
16、在一种可选的实施方式中,所述基于所述拷贝数,分别对所述目标组合区间内的分区区间进行分类,并统计各个分类的区间数量,包括:
17、将所述拷贝数小于第二预设阈值的分区区间划分为第一类别,统计得到第一区间数量;
18、将所述拷贝数大于或等于所述第二预设阈值、且小于或等于第三预设阈值的分区区间划分为第二类别,统计得到第二区间数量;
19、将所述拷贝数大于所述第三预设阈值的分区区间划分为第三类别,统计得到第三区间数量;
20、其中,第二预设阈值小于第三预设阈值。
21、在一种可选的实施方式中,所述第二预设阈值的取值范围为1.6-1.8,所述第三预设阈值的取值范围为2.2-2.4。
22、在一种可选的实施方式中,所述基于所述目标组合区间的多个所述区间数量,确定对应的分区区间筛选规则,包括:
23、若所述第一区间数量和所述第三区间数量之和,小于或等于所述第二区间数量,则确定对应的分区区间筛选规则为:将所述目标组合区间中拷贝数在第一预设范围内的分区区间筛选出来;
24、若所述第二区间数量和所述第三区间数量之和,小于或等于所述第一区间数量,则确定对应的分区区间筛选规则为:将所述目标组合区间中拷贝数在第二预设范围内的分区区间筛选出来;
25、若所述第一区间数量和所述第二区间数量之和,小于或等于所述第三区间数量,则确定对应的分区区间筛选规则为:将所述目标组合区间中拷贝数在第二预设范围内的分区区间筛选出来;
26、其中,所述第一预设范围是基于所述第二预设阈值和所述第三预设阈值设定的拷贝数范围;所述第二预设范围是基于所述目标组合区间的拷贝数设定的拷贝数范围。
27、在一种可选的实施方式中,所述第一预设范围为1.9-2.1;和/或,所述第二预设范围的起点值为所述目标组合区间的拷贝数减去0.1,所述第二预设范围的终点值为所述目标组合区间的拷贝数加上0.1。
28、在一种可选的实施方式中,所述获取基因组序列样本数据各个分区区间的拷贝数,包括:
29、获取所述基因组序列样本数据各个分区区间的拷贝数比率;
30、若所述基因组序列样本数据中包括y染色体数据,则对于y染色体对应的第二分区区间和x染色体对应的第二分区区间,确定拷贝数为;对于所述第二分区区间以外的其他分区区间,确定拷贝数为;
31、若所述基因组序列样本数据中不包括y染色体数据,则各个所述分区区间的拷贝数确定为;
32、其中,ratio为拷贝数比率。
33、第二方面,本发明提供了一种基于超低深度全基因组高通量测序的基因组拷贝数变异分析数据的处理装置,该装置包括:
34、区间拷贝数获取模块,用于获取基因组序列样本数据各个分区区间的拷贝数;
35、第一筛选模块,用于对于y染色体对应的第一分区区间,若拷贝数小于第一预设阈值,则删除所述第一分区区间;
36、合并模块,用于合并拷贝数相似的相邻分区区间,形成组合区间;
37、第二筛选模块,用于按照区间大小对所述组合区间进行筛选,得到大于预设阈值的目标组合区间;
38、分类统计模块,用于基于所述拷贝数,分别对所述目标组合区间内的分区区间进行分类,并统计各个分类的区间数量;
39、第三筛选模块,用于针对每个所述目标组合区间,基于所述目标组合区间对应的多个所述区间数量,对所述目标组合区间中的分区区间进行筛选,得到目标分区区间;
40、绘图显示模块,用于基于所述目标分区区间中的数据进行绘图显示。
41、第三方面,本发明提供了一种计算机设备,包括:存储器和处理器,存储器和处理器之间互相通信连接,存储器中存储有计算机指令,处理器通过执行计算机指令,从而执行上述第一方面或其对应的任一实施方式的基因组拷贝数变异分析数据的处理方法。
42、第四方面,本发明提供了一种计算机可读存储介质,该计算机可读存储介质上存储有计算机指令,计算机指令用于使计算机执行上述第一方面或其对应的任一实施方式的基因组拷贝数变异分析数据的处理方法。
43、第五方面,本发明提供了一种计算机程序产品,包括计算机指令,计算机指令用于使计算机执行上述第一方面或其对应的任一实施方式的基因组拷贝数变异分析数据的处理方法。
44、针对低深度、甚至超低深度(0.1×)全基因组的cnv结果图主要表现为离散的点图,给辨别cnv的实际情况带来挑战的问题,本实施例提出了基于超低深度全基因组高通量测序的基因组拷贝数变异分析数据的处理方法、装置及设备,提供了能够准确呈现全基因组cnv结果的数据筛选逻辑和条件,排除干扰信息,减少争议位点,有利于结果的直观呈现,减少结果解读的争议,便于正确解读,提高了检测结果的准确性和可读性。
45、另外,本发明实施例中,通过删除拷贝数较小的y染色体对应的第一分区区间,排除了试剂、测序错误等因素带来的误差干扰,进一步提高了检测结果的准确性和可读性。
- 还没有人留言评论。精彩留言会获得点赞!