一种光动力型可充电抗菌抗病毒纳米纤维膜的制备方法
1.本发明属于纤维膜技术领域,具体是一种光动力型可充电抗菌抗病毒纳米纤维膜的制备方法。
背景技术:
2.传染病是由各种各样的病原体所引起的,可以在人与人、动物与动物或人与动物之间相互传播的一种疾病。虽然医护人员穿戴的个人防护设备可以显著减少病原体的传播,但感染风险不能完全消除。就目前的抗菌材料来说,其消耗不可逆、释放周期短、长期使用后具有诱导细菌耐药性的风险。
3.光动力技术通过可见光照射产生活性氧(ros)与细菌上的靶点结合,从而抑制蛋白质的合成,造成细菌的溶解与死亡。考虑到细菌和病毒的遗传灵活性,这种多靶点的灭活方式使得光动力疗法具有广谱的灭杀生物活性从而避免细菌和病毒产生耐药性。文献《daylight-driven photosensitive antibacterial melt-blown membranes for medical use》将对苯二甲酰二邻苯二甲酸和表没食子儿茶素没食子酸酯接枝到经过亲水改性的ppcl熔喷膜上,但该研究的ros产量较低。因此,制备一种既可以在光照和黑暗条件下工作,又可充电且ros释放量高的光动力型抗菌抗病毒材料来解决现有问题十分重要。
技术实现要素:
4.针对现有技术的不足,本发明拟解决的技术问题是,提供一种光动力型可充电抗菌抗病毒纳米纤维膜的制备方法。
5.本发明解决所述技术问题的技术方案是,提供一种光动力型可充电抗菌抗病毒纳米纤维膜的制备方法,其特征在于,该方法包括以下步骤:
6.1)将纤维膜浸入pda溶液中,pda自聚沉积到纤维膜的纤维上;沉积完成后取出,洗涤除杂后,再干燥去除洗涤剂,得到pan@pda纳米纤维膜;
7.2)将tdpa与多聚磷酸溶解在溶剂中,得到均相的tdpa/多聚磷酸溶液;再将pan@pda纳米纤维膜浸泡在tdpa/多聚磷酸溶液中进行接枝反应,tdpa通过酯化反应接枝到pan@pda纳米纤维膜的纤维上;反应完成后取出,洗涤除杂后,再干燥去除洗涤剂,得到pan@pda/tdpa纳米纤维膜;
8.3)将ca与多聚磷酸溶解在溶剂中,得到均相的ca/多聚磷酸溶液;再将pan@pda/tdpa纳米纤维膜浸泡在ca/多聚磷酸溶液中进行接枝反应,ca通过酯化反应接枝到pan@pda/tdpa纳米纤维膜的tdpa上;反应完成后取出,洗涤除杂后,再干燥去除洗涤剂,得到光动力型可充电抗菌抗病毒纳米纤维膜。
9.与现有技术相比,本发明的有益效果在于:
10.(1)本发明采用纤维膜作为基膜,通过浸渍的方式将光敏剂tdpa和抗菌剂ca接枝到pan@pda纳米纤维膜上,形成复合抗菌剂,克服单一抗菌剂效果差的缺点,得到可充电、可储存和高效持久抗菌抗病毒的纳米纤维膜。
11.(2)tdpa中含有双二苯甲酮结构在吸收能量后可以释放活性氧,具有抗菌和抗病毒性能,并通过氢夺取和氧猝灭的方式重新还原为二苯甲酮,从而解决抗菌材料消耗不可逆、释放周期短、长期使用后具有诱导细菌耐药性等问题,实现高效持久的抗菌和抗病毒。
12.(3)本发明制备的纳米纤维膜对大肠杆菌和金黄色葡萄球菌均有优异的灭杀效果,在光照和黑暗条件下,对大肠杆菌和金黄色葡萄球菌的杀菌率均达95%以上。纳米纤维膜具有可充电、可储存性能,经过7个循环测试后充电容量保留了原来的65%~75%。纳米纤维膜具有结构稳定性,lat结构稳定性为65%~70%。纳米纤维膜的
·
oh和h2o2的量分别为5900~6300μg/g和780~850μg/g,具有优异的抗病毒性能。
13.(4)本发明的基膜选材广泛,可采用静电纺丝膜、熔喷膜或sms复合非织造膜。其中,静电纺丝技术制得的pan纳米纤维膜具有较高比表面积、多孔结构以及具有潜在病原体拦截能力。
附图说明
14.图1为本发明的实施例1及对比例1-4制备的纳米纤维膜的傅里叶红外光谱图;
15.图2为本发明的实施例1和对比例1-4制备的纳米纤维膜的透湿性测试;
16.图3为本发明的实施例1及对比例3-4制备的纳米纤维膜在日光照射1h后,在黑暗条件下羟基自由基(
·
oh)的释放量图;
17.图4为本发明的实施例1及对比例3-4制备的纳米纤维膜在日光照射1h后,在黑暗条件下过氧化氢(h2o2)的释放量图;
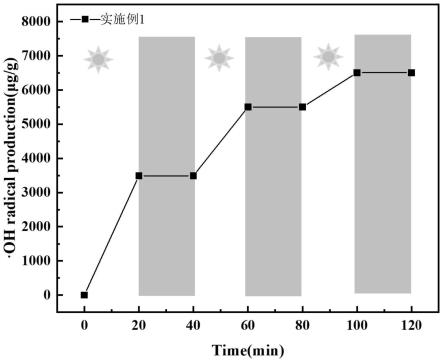
18.图5为本发明的实施例1制备的纳米纤维膜的羟基自由基的释放量与时间的关系图;
19.图6为本发明的实施例1制备的纳米纤维膜的过氧化氢的释放量与时间的关系图;
20.图7为本发明的实施例1制备的纳米纤维膜的可充电循环测试图;
21.图8为本发明的实施例1制备的纳米纤维膜的lat结构随储存时间的变化图;
22.图9为本发明的实施例1制备的纳米纤维膜在光照(light(+))条件下与大肠杆菌(e.coli)共培养1h后的菌落情况;
23.图10为本发明的实施例1制备的纳米纤维膜在光照条件下与金黄色葡萄球菌(s.aureus)共培养1h后的菌落情况;
24.图11为本发明的实施例1制备的纳米纤维膜在黑暗(light(-))条件下与大肠杆菌共培养1h后的菌落情况;
25.图12为本发明的实施例1制备的纳米纤维膜在黑暗条件下与金黄色葡萄球菌共培养1h后的菌落情况;
26.图13为本发明的实施例1制备的纳米纤维膜在光照条件下分别与e.coli和s.aureus共培养0min、10min、30min、60min后的杀菌率测试图;
27.图14为本发明的实施例1制备的纳米纤维膜在黑暗条件下分别与e.coli和s.aureus共培养0min、10min、30min、60min后的杀菌率测试图;
28.图15为本发明的对比例1和实施例1的抗病毒性能图。
具体实施方式
29.本发明提供了一种光动力型可充电抗菌抗病毒纳米纤维膜的制备方法(简称方法),其特征在于,该方法包括以下步骤:
30.1)将纤维膜浸入pda(聚多巴胺)溶液中,pda自聚沉积到纤维膜的纤维上;沉积完成后取出,洗涤除杂后,再干燥去除洗涤剂,得到pan@pda纳米纤维膜;
31.优选地,步骤1)中,纤维膜采用静电纺丝膜、熔喷膜或sms复合非织造膜;静电纺丝膜的聚合物材质选用pan(聚丙烯腈)、pva(聚乙烯醇)、pcl(聚己内酯)、pvp(聚乙烯吡咯烷酮)、tpu(热塑性聚氨酯弹性体橡胶)或pva-co-pe(聚乙烯醇共聚乙烯);熔喷膜的聚合物材质选用pp(聚丙烯)、pp/pcl复配(聚丙烯/聚己内酯)、ptfe/pp复配(聚四氟乙烯/聚丙烯)或pp/pc复配(聚丙烯/聚碳酸脂);sms复合非织造膜的聚合物材质选用pp。
32.优选地,步骤1)中,静电纺丝的具体工艺是:将聚合物溶液吸入注射器中,在15~20kv的电压下用注射泵控制聚合物溶液的挤出流量,将静电纺丝形成的纤维膜收集在覆盖有离型纸的金属滚筒上,得到聚合物纳米纤维膜;注射泵的速度为1~2ml/h,收集时间为5~7h,滚筒转速为60~100rpm。
33.优选地,步骤1)中,pan静电纺丝膜的制备是:将pan粉末溶解于pan的良溶剂中,得到均相的pan溶液作为纺丝液,再通过静电纺丝法得到pan纳米纤维膜;pan溶液的浓度为8~14wt%(优选10~12wt%);pan溶解的温度为30~60℃,采用搅拌溶解,搅拌时间为3~7h。所述pan的良溶剂可采用n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)或二甲基亚砜(dmso)。
34.优选地,步骤1)中,配置pda溶液具体是:将盐酸多巴胺和三羟甲基氨基甲烷盐酸盐溶解在去离子水中,再将3-氨丙基三乙氧基硅烷和无水乙醇混合后倒入,搅拌得到pda溶液。
35.优选地,步骤1)中,pda溶液中,盐酸多巴胺的质量、三羟甲基氨基甲烷盐酸盐的质量、去离子水的体积、3-氨丙基三乙氧基硅烷的质量与无水乙醇的体积之比为0.1~0.4g:0.06~0.24g:50~200ml:0.2~0.8g:10~40ml;自聚沉积的温度为室温,时间为8~16h。
36.优选地,步骤1)中,干燥温度为40~60℃,干燥时间为2~6h。
37.优选地,步骤1)中,洗涤除杂采用洗涤剂a洗去pda溶液;洗涤剂a采用去离子水。
38.2)将tdpa(4,4
’‑
对苯二甲酰二邻苯二甲酸酐)与多聚磷酸溶解在溶剂中,得到均相的tdpa/多聚磷酸溶液;再将pan@pda纳米纤维膜浸泡在tdpa/多聚磷酸溶液中进行接枝反应,tdpa通过酯化反应接枝到pan@pda纳米纤维膜的纤维上;反应完成后取出,洗涤除杂后,再干燥去除洗涤剂,得到pan@pda/tdpa纳米纤维膜;
39.优选地,步骤2)中,tdpa的质量、多聚磷酸的质量与溶剂的体积比为0.1~0.4g:0.1~0.4g:10~40ml;接枝反应过程中持续搅拌,接枝温度为30~70℃,接枝时间为1~3h。
40.优选地,步骤2)中,干燥采用真空烘干,温度为30~50℃,时间为1~3h。
41.3)将ca(天然多酚绿原酸)与多聚磷酸溶解在溶剂中,得到均相的ca/多聚磷酸溶液;再将pan@pda/tdpa纳米纤维膜浸泡在ca/多聚磷酸溶液中进行接枝反应,ca通过酯化反应接枝到pan@pda/tdpa纳米纤维膜的tdpa上;反应完成后取出,洗涤除杂后,再干燥去除洗涤剂,得到pan@pda/tdpa/ca纳米纤维膜,即光动力型可充电抗菌抗病毒纳米纤维膜(简称纳米纤维膜)。
42.优选地,步骤3)中,ca的质量、多聚磷酸的质量与溶剂的体积比为0.1~0.4g:0.1~0.4g:10~40ml;接枝反应过程中持续搅拌,接枝温度为30~70℃,接枝时间为1~3h。
43.优选地,步骤2)和步骤3)中,溶剂为tdpa、ca和多聚磷酸的良溶剂,具体是dmf、四氢呋喃(thf)或1,4-二氧六环。
44.优选地,步骤2)中,洗涤除杂采用洗涤剂b洗去多聚磷酸、溶剂和未反应的tdpa;步骤3)中,洗涤除杂采用洗涤剂b洗去多聚磷酸、溶剂和未反应的ca;洗涤剂b选用丙酮或乙醇。
45.优选地,步骤3)中,干燥采用真空烘干,温度为40~60℃,时间为1~3h。
46.本发明制得的光动力型可充电抗菌抗病毒纳米纤维膜可用于防护材料。
47.下面给出本发明的具体实施例。具体实施例仅用于进一步详细说明本发明,不限制本发明权利要求的保护范围。
48.实施例1
49.(1)将2g的pan粉末溶解在14.67ml的dmf中搅拌6h,搅拌温度为45℃,充分混合均匀得到浓度为12wt%的pan溶液;将pan溶液吸入10ml注射器中,在15kv的电压下用注射泵以2ml/h的速度控制pan溶液的挤出流量,将静电纺丝形成的纤维膜收集在覆盖有离型纸的金属滚筒上,得到pan纳米纤维膜;
50.将0.2g盐酸多巴胺和0.12g三羟甲基氨基甲烷盐酸盐溶解在100ml的去离子水中,再将0.4g的3-氨丙基三乙氧基硅烷和20ml的无水乙醇混合后倒入,搅拌得到pda溶液;
51.然后将pan纳米纤维膜浸入pda溶液中,在室温下自聚沉积12h;沉积完成后取出,用去离子水洗涤,再干燥4h,得到pan@pda纳米纤维膜;
52.(2)将0.2g的tdpa与0.2g多聚磷酸溶解在20ml的1,4-二氧六环中,得到均相的tdpa/多聚磷酸溶液;再将0.1g的pan@pda纳米纤维膜浸泡在tdpa/多聚磷酸溶液中进行接枝反应,60℃搅拌反应2h;反应完成后取出,用丙酮洗涤,再在40℃的真空烘箱中干燥2h,得到pan@pda/tdpa纳米纤维膜;
53.(3)将0.2g的ca与0.2g多聚磷酸溶解在20ml的1,4-二氧六环中,得到均相的ca/多聚磷酸溶液;再将0.1g的pan@pda/tdpa纳米纤维膜浸泡在ca/多聚磷酸溶液中进行接枝反应,60℃搅拌反应2h;反应完成后取出,用丙酮洗涤,再在40℃的真空烘箱中干燥2h,得到pan@pda/tdpa/ca纳米纤维膜。
54.图5中,白色表示照射、灰色表示黑暗,对实施例1进行20min间隔明暗疲劳测试。由图5可以看出,
·
oh在光照时产生,在黑暗时停止。但在此过程中实施例1的活性并没有降低,每次循环后ros的释放量依然稳定增大。
55.图6中,白色表示照射、灰色表示黑暗,对实施例1进行20min间隔明暗疲劳测试。由图6可以看出,h2o2在光照时产生,在黑暗时停止。但在此过程中实施例1的活性并没有降低,每次循环后ros的释放量依然稳定增大。
56.图7通过反复充电和猝灭七个循环来评估实施例1制备的纳米纤维膜的可充电性。由图10可以看出,在每个循环中先将实施例1光照1h,测量完ros的释放量后用过量的硫代硫酸盐溶液猝灭。经过七个循环后,充电容量没有明显的下降,分别保留了原来充电容量的70.32%和71.28%,表明该纳米纤维膜可以重复多次利用。
57.由图8可以看出,lat结构相关的吸收峰表现出缓慢的衰减。经测试,实施例1仍保
留了原始lat结构的67%以上,从而突出了结构的稳定性。
58.由图9可以看出,光照条件下,实施例1与e.coli共培养30min时菌落数目与空白菌落数目相比下降明显,共培养1h后几乎没有菌落出现,实施例1显示出较高的杀菌活性。
59.由图10可以看出,光照条件下,实施例1与s.aureus共培养30min时菌落数目与空白菌落数目相比下降明显,共培养1h后几乎没有菌落出现,实施例1显示出较高的杀菌活性。
60.由图11可以看出,黑暗条件下,实施例1与e.coli共培养30min时菌落数目与空白菌落数目相比下降明显,共培养1h后菌落数目较少,实施例1显示出较高的杀菌活性。
61.由图12可以看出,黑暗条件下,实施例1与s.aureus共培养30min时菌落数目与空白菌落数目相比下降明显,共培养1h后菌落数目较少,实施例1显示出较高的杀菌活性。
62.由图13可以看出,实施例1在相同的接触时间内对金黄色葡萄球菌的杀菌率大于大肠杆菌。主要是因为大肠杆菌和金黄色葡萄球菌的细胞壁结构存在差异,进而影响细菌对ros的敏感度。当实施例1与细菌共培养60min时,在光照条件下的杀菌率可以达到99%以上。
63.由图14可以看出,当实施例1与细菌共培养60min时,黑暗条件下的杀菌率可以达到99%以上。
64.根据病毒的基本结构,ros与病毒主要有三个靶点:核酸、病毒蛋白质和病毒脂质。为了评估实施例1的抗病毒性能,选用h3n2流感病毒对实施例1进行攻击。由图15可以看出,实施例1的抗病毒活性值可以达到3.6,这主要归因于实施例1产生的ros对h3n2的rna和脂质膜起到氧化作用。此外,ros与病毒的蛋白质衣壳交联,导致病毒与宿主细胞的特定结合位点失活。
65.对比例1
66.采用实施例1的步骤(1)得到的pan纳米纤维膜。
67.对比例2
68.采用实施例1的步骤(1)得到的pan@pda纳米纤维膜。
69.对比例3
70.将0.2g的ca和5g的cdi(n,n-羰基二咪唑)溶于20ml的thf中,得到均相的ca/cdi溶液;再将0.1g的实施例1的步骤(1)的pan@pda纳米纤维膜浸泡在ca/cdi溶液中进行接枝反应,60℃搅拌反应2h;反应完成后取出,用丙酮洗涤,再在40℃的真空烘箱中干燥2h,得到pan@pda/ca纳米纤维膜。
71.对比例4
72.采用实施例1的步骤(2)得到的pan@pda/tdpa纳米纤维膜。
73.由图1可以看出,所有样品在3647、3564、2932、2865、2241、1732、1665、1451cm-1
处都出现了pan的特征吸收峰。对比例2在3679-2938、1614、1519cm-1
处出现新的特征峰,主要归因于pda的o-h、n-h和c-nh基团拉伸振动。当pda原位生长在pan纳米纤维膜上后,pan的特征峰没有发生改变,说明聚多巴胺的原位生长是物理现象。由于实施例1和对比例3-4都是经过pda改性的,所以在这三个例子的纤维膜上pda的吸收峰没有发生改变。其中501cm-1
、773cm-1
、1114-1455cm-1
处峰值的振动说明实施例1和对比例3-4不是物理现象而是化学接枝,表明成功接枝。
74.由图2可以看出,实施例1和对比例1-4的透湿性之间存在一定的差异,主要是因为纳米纤维膜经过亲水性的聚多巴胺处理后,其中含有的亲水基团对水分子的吸引力增大。所以经过改性后的纳米纤维膜表现出良好的透湿性能。
75.由图3可以看出,在最初的5min内,实施例1释放
·
oh的速率最大,随着时间的增加,ros的浓度缓慢增加,最后呈现出饱和状态。通过测试可以看出实施例1最大释放出
·
oh的量为6188.56μg/g。对应于103.14μg/g/min的充电速率,从而表示大量的光能可以被纳米纤维膜充分利用。
76.由图4可以看出,在最初的5min内,实施例1释放h2o2的速率最大,随着时间的增加,ros的浓度缓慢增加,最后呈现出饱和状态。通过测试可以看出实施例1最大释放出h2o2的量842.00μg/g。对应于14.03μg/g/min的充电速率,从而表示大量的光能可以被纳米纤维膜充分利用。
77.实施例2
78.(1)将1.63g的pan粉末溶解在14.67ml的dmf中搅拌6h,搅拌温度为45℃,充分混合均匀得到浓度为10wt%的pan溶液;将pan溶液吸入10ml注射器中,在20kv的电压下用注射泵以2ml/h的速度控制pan溶液的挤出流量,将静电纺丝形成的纤维膜收集在覆盖有离型纸的金属滚筒上,得到pan纳米纤维膜;
79.将0.2g盐酸多巴胺和0.12g三羟甲基氨基甲烷盐酸盐溶解在100ml的去离子水中,再将0.4g的3-氨丙基三乙氧基硅烷和20ml的无水乙醇混合后倒入,搅拌得到pda溶液;
80.然后将pan纳米纤维膜浸入pda溶液中,在室温下自聚沉积8h;沉积完成后取出,用去离子水洗涤,再干燥4h,得到pan@pda纳米纤维膜;
81.(2)将0.2g的tdpa与0.2g多聚磷酸溶解在20ml的1,4-二氧六环中,得到均相的tdpa/多聚磷酸溶液;再将0.1g的pan@pda纳米纤维膜浸泡在tdpa/多聚磷酸溶液中进行接枝反应,40℃搅拌反应3h;反应完成后取出,用丙酮洗涤,再在40℃的真空烘箱中干燥2h,得到pan@pda/tdpa纳米纤维膜;
82.(3)将0.2g的ca与0.2g多聚磷酸溶解在20ml的1,4-二氧六环中,得到均相的ca/多聚磷酸溶液;再将0.1g的pan@pda/tdpa纳米纤维膜浸泡在ca/多聚磷酸溶液中进行接枝反应,70℃搅拌反应1h;反应完成后取出,用丙酮洗涤,再在40℃的真空烘箱中干燥2h,得到pan@pda/tdpa/ca纳米纤维膜。
83.实施例3
84.(1)将2.39g的pan粉末溶解在14.67ml的dmf中搅拌6h,搅拌温度为45℃,充分混合均匀得到浓度为14wt%的pan溶液;将pan溶液吸入10ml注射器中,在17kv的电压下用注射泵以2ml/h的速度控制pan溶液的挤出流量,将静电纺丝形成的纤维膜收集在覆盖有离型纸的金属滚筒上,得到pan纳米纤维膜;
85.将0.2g盐酸多巴胺和0.12g三羟甲基氨基甲烷盐酸盐溶解在100ml的去离子水中,再将0.4g的3-氨丙基三乙氧基硅烷和20ml的无水乙醇混合后倒入,搅拌得到pda溶液;
86.然后将pan纳米纤维膜浸入pda溶液中,在室温下自聚沉积16h;沉积完成后取出,用去离子水洗涤,再干燥4h,得到pan@pda纳米纤维膜;
87.(2)将0.2g的tdpa与0.2g多聚磷酸溶解在20ml的1,4-二氧六环中,得到均相的tdpa/多聚磷酸溶液;再将0.1g的pan@pda纳米纤维膜浸泡在tdpa/多聚磷酸溶液中进行接
枝反应,70℃搅拌反应1h;反应完成后取出,用丙酮洗涤,再在40℃的真空烘箱中干燥2h,得到pan@pda/tdpa纳米纤维膜;
88.(3)将0.2g的ca与0.2g多聚磷酸溶解在20ml的1,4-二氧六环中,得到均相的ca/多聚磷酸溶液;再将0.1g的pan@pda/tdpa纳米纤维膜浸泡在ca/多聚磷酸溶液中进行接枝反应,40℃搅拌反应3h;反应完成后取出,用丙酮洗涤,再在40℃的真空烘箱中干燥2h,得到pan@pda/tdpa/ca纳米纤维膜。
89.本发明未述及之处适用于现有技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1