靶基因组区域的灵活且高通量的测序的制作方法
靶基因组区域的灵活且高通量的测序
1.相关申请的交叉引用
2.本技术要求于2019年5月30日提交的美国临时申请62/854,458的权益,其公开内容通过引用整体(包括所有图、表和氨基酸或核酸序列)并入本文。
3.此申请的序列表标记为“seq-list.txt”,创建于2020年5月15日,大小为2kb。序列表的全部内容通过引用整体并入本文。
背景技术:
4.随着更强大且负担得起的测序技术的出现,靶向测序变得越来越重要。大多数用于分析基因组目标区域的常规方法涉及目标杂交和捕获(gnirke等,2009)、多重pcr(campbell等,2015)或分子倒置探针(shen等,2011)。这些方法要么昂贵、难以优化、数据可变性高,要么对不同长度的序列目标缺乏灵活性。因此,需要改进的方法用于分析,例如检测和测序靶基因组区域,特别是检测和测序包含或预期包含遗传多态性的靶基因组区域。
技术实现要素:
5.本文公开的某些实施方式提供了用于捕获靶基因组区域和任选地例如通过检测和/或测序进一步分析靶基因组区域的材料和方法。优选地,靶基因组区域包含或预期包含遗传多态性。
6.在某些实施方式中,本文公开的用于从目标遗传物质捕获靶基因组区域的方法包括将延伸探针和连接探针(ligation probe)与第一靶序列和第二靶序列杂交,其中第一靶序列和第二靶序列位于靶基因组区域两侧;将延伸探针的3'端延长,直到延长的延伸探针的3'端与连接探针的5'端相邻;将延长的延伸探针的3'端与连接探针的5'端连接以产生扎结探针(ligated probe),扎结探针包含靶基因组区域,从而捕获靶基因组区域。
7.扎结探针可以任选地从反应混合物中纯化,并用扩增引物对进行pcr扩增,以产生适合进一步检测和/或测序的扎结探针的双链拷贝。可以使用下一代测序技术进行测序,例如纳米孔测序、可逆染料终止子测序、单分子实时(smrt)测序或双端测序。
8.本发明的进一步实施方式提供了产生双链形式的延伸和连接探针的方法。使用双链形式的探针允许捕获双链靶基因组区域的两条链。
9.在某些实施方式中,使用多对探针捕获遗传物质中的多个靶基因组区域,每对探针包含延伸探针和连接探针,扩增与相应靶序列杂交的延伸探针并连接扩增的延伸探针与相应的连接探针以捕获多个靶基因组区域。扎结探针可以任选地从反应混合物中纯化,并用扩增引物对进行pcr扩增以产生适合进一步检测和测序的靶基因组区域的双链拷贝。来自多个样品的多个扎结探针可以合并以在多重测序反应中测序。扩增引物可包含独特的标识符序列,以鉴定扩增的靶基因组区域的来源。在测序步骤之后,样品特定的唯一标识符用于将一个序列分配给一个样品,将捕获的靶基因组区域的序列与已知数据库进行比较,以将序列分配给样品中的靶基因组区域。可以使用下一代测序技术进行测序,例如纳米孔测序、可逆染料终止子测序、单分子实时(smrt)测序或双端测序。
10.本发明的进一步实施方式提供了用于实施本文公开的方法的试剂盒。所述试剂盒包括以下各项中的一项或多项:一对或多对延伸探针和连接探针、酶(例如dna连接酶、dna聚合酶)、一对或多对扩增引物、测序试剂和进行测定的说明书。
附图说明
11.本专利或申请文件至少包含一张彩色绘图。带有彩色附图的本专利或专利申请公开的拷贝,将在请求和支付必要费用后由专利局提供。
12.图1:根据本文公开的方法捕获和测序靶基因组区域的一个实施例的概述。
13.图2:根据本文公开的方法捕获和测序长靶基因组区域的一个实施例的概述。
14.图3:制备无需用于修饰的尾部交换的双链形式的探针的一个实施例的概述。
15.图4:制备具有尾部交换以包含所需的修饰的双链形式的探针的一个实施例的概述。
16.图5:制备双链形式的上游或下游探针的方法的概述。
17.图6:使用双链上游和下游探针分析靶基因组区域的两条链的方法的一个实施例的概述。双链上游和下游探针(例如,分别通过图3或4中例示的方法制备)可用于分析靶基因组区域的两条链。
具体实施方式
18.如本文所用,单数形式“一个”和“该”也旨在包括复数形式,除非上下文另有明确指示。就具体说明书和/或权利要求中使用的术语“包括”、“具有”、“带有”或其变体而言,此类术语旨在以类似于术语“包含”的方式是包容的。过渡性术语/短语(及其任何语法变体)“包含”、“基本上由
……
组成”和“组成”可以互换使用。
19.短语“基本上由
……
组成”表示所描述的实施方式包括包含特定材料或步骤的实施方式以及不实质上影响所描述的实施方式的基本和新颖特征的那些实施方式。
20.术语“约”是指在由本领域普通技术人员确定的针对特定值的可接受误差范围内,这将部分取决于该值是如何测量或确定的,即,测量系统的限制。在使用术语“约”的多核苷酸长度的上下文中,这些多核苷酸包含所述数目的碱基或碱基对,在该值附近有0-10%的变化(x
±
10%)。
21.在本公开中,范围以速记方式陈述,以避免必须详细陈述并描述该范围内的每个值。在适当的情况下,可以选择该范围内的任何合适的值作为该范围的上限值、下限值或终点。例如,0.1-1.0的范围代表0.1和1.0的终点,以及0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9的中间值,以及包含在0.1-1.0内的所有中间范围,例如0.2-0.5、0.2-0.8、0.7-1.0等。可以设想一个范围内至少具有两位有效数字的值,例如5-10的范围表示5.0和10.0之间以及5.00到10.00之间的所有值,包括终端值。当范围在本文中使用时,例如对于多核苷酸的大小,范围的组合和子组合(例如,所公开范围内的亚范围)和其中的具体实施方式被明确包括。
22.如本文所用,术语“生物体”包括病毒、细菌、真菌、植物和动物。生物体的其他实施例是本领域普通技术人员已知的,并且此类实施方式在本文公开的材料和方法的范围内。本文所述的测定可用于分析从任何生物体获得的任何遗传物质。
23.如本文所用,术语“基因组”、“基因组的”、“遗传物质”或它们的其他语法变体是指
来自任何生物体的遗传物质。遗传物质可以是病毒基因组dna或rna、核遗传物质(例如基因组dna)或存在于细胞器中的遗传物质(例如线粒体dna或叶绿体dna)。它还可以代表来自天然或人工混合物的遗传物质,或来自几种生物的遗传物质的混合物。
24.如本文所用,“靶基因组区域”是生物体遗传物质中的感兴趣区域。
25.术语“与...杂交”在用于两个序列时表示两个序列彼此充分互补以允许两个序列之间的核苷酸碱基配对。彼此杂交的序列可以完全互补,但也可能在一定程度上存在错配。因此,本文所述的延伸和连接探针的5'和3'端的序列可能与靶基因组区域的5'和3'端的相应靶序列存在一些错配,只要延伸和连接探针可以与靶序列杂交以促进靶基因组区域的捕获即可。根据杂交的严格度,两个互补序列之间高达约5%至20%的错配将允许两个序列之间的杂交。通常,高严格度条件具有较高的温度和较低的盐浓度,而低严格度条件具有较低的温度和较高的盐浓度。优选杂交的高严格度条件,因此,延伸和连接探针3'和5'末端的序列优选与靶基因组区域3'和5'末端的相应靶序列完美互补。
26.如本文所用,术语“标识符”是指4至100个核苷酸之间、优选地10至20个核苷酸之间以及甚至更优选地约8个或16个核苷酸之间的已知核苷酸序列。标识符序列的适当长度取决于所使用的测序技术。一旦并入扩增的靶基因组区域中,标识符序列可以促进靶基因组区域的测序和鉴定,例如,通过提供允许将正确序列分配到正确靶基因组区域的独特识别位点。
27.本文中使用的术语“双端测序”是指使用存在于双链多核苷酸每一端的特异性引物结合位点对双链多核苷酸的两端进行测序的测序技术。双端测序生成高质量的测序数据,其使用计算机软件程序进行对齐,以生成两侧为两个引物结合位点的多核苷酸序列。从双链分子的两端进行测序可以获得来自双链分子两端的高质量数据,因为仅从分子的一端进行测序可能会导致测序质量随着执行更长的测序读取而下降。
28.在双端测序中,在本文公开的方法的pcr扩增步骤结束时产生的双链扩增的扎结探针使用结合双链扎结探针两端的特异性引物进行测序。双端测序的一般描述和原理在“illumina sequencing technology,illumina,公开号770-2007-002”中提供,其内容通过引用整体并入本文。
29.双端测序技术的非限制性实施例由illuminamiseq
tm
、illuminamiseqdx
tm
和illuminamiseqfgx
tm
提供。可用于本文公开的测定中的双端测序技术的其他实施例是本领域已知的,此类实施方式在本发明的范围内。
30.如本文所用,短语“发夹接头”是指包含双链主干和单链发夹环的多核苷酸。发夹接头的单链发夹环区域可为测序提供引物结合位点。因此,一旦发夹接头与靶基因组序列的两个粘性末端杂交,它就会产生一个双链dna模板,该模板在两端由发夹环封端的双链区域中包含靶基因组区域。此类模板可用于通过单分子实时(smrt)测序(pacbio
tm
)对靶基因组区域进行测序。“pacificbiosciences(2018),公开号br108-100318”中提供了smrt测序的描述和原理,其内容通过引用整体并入本文。
31.纳米孔技术可用于本文公开的方法中,以对靶基因组区域进行测序。在某些此类实施方式中,如例如在“nanopore technology brochure,oxford nanopore technologies(2019)”和“nanopore product brochure,oxford nanopore technologies(2018)”中所述,处理靶基因组区域的拷贝以对靶基因组区域进行测序。这两本手册的内容通过引用整
体并入本文。
32.在本公开中,不同的序列由特定的命名法描述,例如引物结合序列、引物序列、标识符序列、测序引物结合序列和测序引物序列。当使用这样的命名法时,应理解所识别的序列与相应序列的至少一部分基本上相同或基本上反向互补。例如,“引物序列”描述与引物序列的至少一部分基本相同或与引物序列的至少一部分基本反向互补的序列。这是因为,当捕获的靶基因组区域被转化为包含引物结合序列的双链形式时,双链靶基因组区域可以使用具有与引物结合序列的至少一部分基本相同或基本反向互补的序列的引物进行测序。因此,此处使用命名法,以简化本文公开的方法中使用的不同多核苷酸和多核苷酸部分的描述;然而,本领域普通技术人员会认识到,可以使用适当的与相应序列的至少一部分基本相同或基本反向互补的适当序列来实践本文公开的方法。
33.此外,彼此对应的两个序列,例如引物结合序列和引物序列,或测序引物结合序列和测序引物序列,在至少70%、优选地至少80%、甚至更优选地至少90%、并且最优选地至少95%的序列上,具有至少90%的序列同一性,优选至少95%的序列同一性,甚至更优选地至少97%的序列同一性,并且最优选地至少99%的序列同一性。或者,彼此对应的两个序列彼此反向互补,并在至少70%、优选地至少80%、甚至更优选地至少90%、并且最优选地至少95%的序列上,具有至少90%的完美匹配,优选地至少95%的完美匹配,甚至更优选地至少97%的完美匹配,最优选地至少99%的完美匹配。因此,彼此对应的两个序列可以彼此杂交,或在至少70%、优选地至少80%、甚至更优选地至少90%、并且最优选地至少95%的序列上与共同参考序列杂交。优选地,彼此对应的两个序列在两个序列的整个长度上100%相同或在两个序列的整个长度上100%反向互补。
34.本公开提供解决与用于分析靶基因组区域的常规方法相关的问题的材料和方法。特别地,本公开提供了用于分析靶基因组区域、特别是具有或怀疑具有遗传多态性的靶基因组区域的材料和方法。
35.本文公开的方法提供从目标遗传物质捕获靶基因组区域。该方法包括以下步骤:
36.a)将延伸探针和连接探针与第一靶序列和第二靶序列杂交,其中第一靶序列和第二靶序列位于靶基因组区域的两侧,其中:
37.i)延伸探针包含朝向3'端的第一靶结合序列和朝向5'端的第一引物结合序列,和
38.ii)连接探针包含朝向5'端的第二靶结合序列和朝向3'端的第二引物结合序列;
39.b)扩增延伸探针的3'端,直到扩增的延伸探针的3'端与连接探针的5'端相邻;
40.c)将扩增的延伸探针的3'端与连接探针的5'端连接以产生扎结探针,扎结探针从5'端到3'端包含第一引物结合序列、第一靶结合序列、扩增的靶基因组区域、第二靶结合序列和第二引物结合序列。
41.延伸探针朝向3'端包含与第一靶序列杂交的序列。延伸探针上的此类序列在本文中称为第一靶结合序列。延伸探针朝向5'端包含第一引物结合序列。第一靶结合序列和第一引物结合序列可以具有可以提供附加功能的间插序列,例如标识符序列。
42.连接探针朝向5'端包含一个与第二靶序列杂交的序列。连接探针上的这种序列在本文中称为第二靶结合序列。连接探针朝向3'端包含第二引物结合序列。第二靶结合序列和第二引物结合序列可具有可提供附加功能的间插序列,例如标识符序列。连接探针的5'端有一个磷酸酯基团,便于连接探针与扩增延伸探针的3'端的连接。
43.因此,本文公开的方法包括将一对专门设计的寡核苷酸探针与目标遗传物质中的某些靶序列杂交的步骤。靶序列位于靶基因组区域的两侧。图1显示了一个包含一个snp的靶基因组区域和与该snp不相邻杂交的探针。第一探针(显示在图1的左侧)在本文中称为“延伸探针”,第二探针(显示在图1的右侧)在本文中称为“连接探针”。延伸探针3'端的序列与遗传物质上相应的靶序列杂交,连接探针5'端的序列与遗传物质上相应的靶序列杂交。因此,延伸探针和连接探针与相应的靶序列结合,并且这些靶序列位于靶基因组区域的两侧。
44.延伸探针和连接探针中的每一个可包含最少约20至约60个核苷酸。特别地,延伸探针的第一靶结合序列部分可以为至少约10至约30个核苷酸。延伸探针的第一引物结合序列也可为至少约10至约30个核苷酸。类似地,连接探针的第二靶序列可以为至少约10至约30个核苷酸,连接探针的第二引物结合序列可以为至少约10至约30个核苷酸。探针对靶结合位点的特异性可由第一和第二靶结合序列的长度控制。特别地,较长长度的第一和第二靶结合序列提供更高的结合特异性,而较短长度的第一和第二靶结合序列提供较低的特异性。本领域普通技术人员可以基于靶基因组区域的序列和特定生物体的可用基因组序列,例如从基因组序列数据库确定第一和第二靶结合序列的合适序列。
45.靶基因组区域的长度以及两个探针的靶序列之间的距离取决于分析的目的、靶基因组区域的特征以及执行时用于分析的测序方法。例如,如果目的是发现靶基因组区域的多态性,例如snp、插入缺失、缺失或插入,则分析约100至约300个碱基对(bp)的靶基因组区域。此外,如果使用illumina
tm
2x150bp测序方法,可分析约300bp的靶基因组区域。如果使用基于双端或纳米孔的测序技术,则可以分析约1000bp至约20000bp的靶基因组区域。或者,如果目的是对snp进行基因分型,则靶基因组区域可以非常短,例如在约10bp和约100bp之间。在本文公开的方法中,靶基因组区域包含至少二至五十个核苷酸。因此,两个探针在目标遗传物质上不相邻地杂交。
46.在杂交步骤结束时,延伸探针通过第一靶结合序列与第一靶序列杂交,连接探针通过第二靶结合序列与第二靶序列杂交。第一和第二靶结合序列位于靶基因组区域的两侧。
47.本文公开的方法的下一步包括延伸反应,以延长延伸探针,即,将延伸探针向连接探针延伸。延伸探针的延伸被设计为填补第一靶序列和第二靶序列之间的间隙,即延伸反应将靶基因组区域的一个序列添加到延伸探针。
48.可以使用缺乏链置换能力的dna聚合酶进行延伸探针的延伸。缺乏链置换能力的dna聚合酶在完全填充第一和第二靶序列之间的间隙时会解离,因此,当它到达连接探针的5'端时会解离。
49.在随后的步骤中,例如在连接酶介导的反应中,将连接探针的5'端连接到延长的延伸探针的3'端。
50.出于本公开的目的并且关于两个探针的结合位点,术语“不相邻”或“不相邻的”表示当两个探针与其相应的靶序列杂交时,延伸探针的3'端不能与连接探针的5'端形成磷酸二酯键。相反,关于两个探针的结合位点,术语“相邻”表示当两个探针与其相应的靶序列杂交时,延伸探针的3'端可以与连接探针的5'端形成磷酸二酯键。
51.因为本文公开的方法涉及在延伸步骤中填充两个探针之间的间隙,所以可以设计
探针以结合靶区域周围任何地方的靶序列,只要这样的靶序列位于靶基因组区域的两侧。因此,扩增步骤为探针设计提供了灵活性,并增加了从靶基因组区域识别多态性的机会。另外,由于填充间隙的步骤,可以根据不具有或不期望具有多态性的序列设计探针,避免了设计多个探针来识别一个多态性,例如单核苷酸多态性(snp)。此外,延长的区域可以捕获多个多态性,并且分析一个靶基因组区域可以提供关于可能存在于两侧是一对探针的靶序列的区域中的多个多态性的信息。
52.在延伸反应结束时,延伸探针利用附加序列延长,延长的延伸探针的3'端与连接探针的5'端相邻。因此,在延伸反应结束时,延长的延伸探针和连接探针是连接反应的底物。
53.因此,本文公开的方法的下一步包括将延长的延伸探针的3'端与连接探针的5'端连接。连接反应可以包括在延长的延伸探针的3'-oh基团和连接探针的5'-磷酸酯基团之间形成磷酸二酯键。因此,两个探针连接在一起。在某些实施方式中,为了为连接反应提供5'-磷酸酯基团,连接探针被设计为具有5'-磷酸酯基团。
54.因此,在连接步骤的某些实施方式中,提供了连接酶,其将延长的延伸探针的3'端与连接探针的5'端共价连接。在优选的实施方式中,连接酶是dna连接酶。dna连接酶是能够催化在一条互补链上的相邻位点结合的两条多核苷酸链(的末端)之间的磷酸二酯键形成的酶。dna连接酶通常需要atp(ec6.5.1.1)或nad(ec6.5.1.2)作为辅因子,来密封双链dna中的切口。可用于连接步骤的dna连接酶包括t4dna连接酶、大肠杆菌dna连接酶、水生栖热菌(taq)连接酶、嗜热栖热菌dna连接酶或火球菌dna连接酶。适用于本文公开的方法的其他的连接酶是本领域已知的,并且此类实施方式在本发明的范围内。
55.延长的延伸探针和连接探针的连接也可以通过延伸和连接探针的3'-oh和5'-磷酸酯基团之间的磷酸二酯键之外的结合来介导。“el-sagheer等(2011),pnas;108(28)11338-11343”描述了某些此类连接。可用于连接连接探针和延伸探针的人工连接的其他实施方式是本领域已知的,并且此类实施方式在本发明的范围内。
56.在本文公开的方法的某些实施方式中,连接步骤之后可以是设计用于从反应混合物中去除不需要的材料的步骤,所述不需要的材料例如是未合并的探针、未连接的延伸产物(例如由探针结合脱靶得到的延伸探针)和靶基因组dna。这一步骤是可选的;然而,当进行时,它大大提高了反应的特异性。
57.在某些实施方式中,使用外切核酸酶去除不需要的材料。如果使用外切核酸酶进行这种去除,则修饰延伸探针和/或连接探针,以保护扎结探针免受外切核酸酶介导的消化。
58.外切核酸酶可对单链和双链核酸具有5'-3'外切核酸酶活性、3'-5'外切核酸酶活性、或5'-3'和3'-5'外切核酸酶活性。可用于本文公开的方法中的外切核酸酶的非限制性实施例包括外切核酸酶i、外切核酸酶iii、外切核酸酶v、外切核酸酶iv、外切核酸酶t、λ外切核酸酶、t7外切核酸酶、链酶(strandase)外切核酸酶和3'-5'外切磷酸二酯酶。本领域普通技术人员可以选择合适的外切核酸酶和延伸和/或连接探针的相应保护。
59.例如,当使用3'-5'外切核酸酶时,朝3'端修饰连接探针。优选地,这种修饰是在3'端的核苷酸上;然而,也可以对不在3'端而是远离3'端的核苷酸进行修饰,以便3'-5'外切核酸酶可以从3'端切割一些核苷酸,但会在修饰的核苷酸处被阻断,因此不能切割整个扎
结探针。
60.或者,当使用5'-3'外切核酸酶时,朝5'端修饰延伸探针。优选地,这种修饰是在5'端的核苷酸上;然而,也可以对不在5'端而是远离5'端的核苷酸进行修饰,这样5'-3'外切核酸酶可能会从5'端切割一些核苷酸,但会在修饰的核苷酸处被被阻断,因此不能切割整个扎结探针。
61.在某些实施方式中,使用具有5'-3'和3'-5'外切核酸酶的外切核酸酶,或使用5'-3'外切核酸酶和3'-5'外切核酸酶的组合。在此类实施方式中,朝5'端修饰延伸探针,而朝3'端修饰连接探针。优选地,延伸探针的这种修饰在5'端的核苷酸上;然而,也可以对不在5'端而是远离5'端的核苷酸进行修饰,这样5'-3'外切核酸酶可能会从5'端切割一些核苷酸,但会在修饰的核苷酸处被阻断,因此不能切割整个扎结探针。类似地,连接探针的这种修饰是在3'端的核苷酸上;然而,也可以对不在3'端而是远离3'端的核苷酸进行修饰,以便3'-5'外切核酸酶可以从3'端切割一些核苷酸,但会在修饰的核苷酸处被阻断,因此不能切割整个扎结探针。
62.本领域普通技术人员可以确定对3'和/或5'端的适当修饰。此类修饰包括在核苷酸之间引入硫代磷酸酯键,向寡核苷酸的5'和/或3'末端结合两个或多个亚磷酰胺和单硫代磷酸酯和/或二硫代磷酸酯键,将相邻核苷酸之间的一个或多个磷酸二酯键替换为甲缩醛/缩酮型键,通过磷酰基或乙酰基封闭3'末端羟基,引入3'末端氨基磷酸酯修饰,引入肽核酸(pna)或锁核酸(lna),引入一个或多个硫代磷酸酯基团,或在寡核苷酸骨架中引入2-o-甲基核糖基团。
63.可用于本文公开的方法的修饰的非限制性实施例公开于:美国专利4,656,127;shaw等1991,nucleic acids research,19,747-750;raney等(1998),peptide nucleic acids(nielsen,p.e.和egholm,m.,eds.),horizon scientific press,wymondham,u.k.;simeonov等,nucl.acids res.2002,vol.30,e31;和jacobsen等int.biot.lab,2001年2月,18。这些参考文献中的每一篇均通过引用整体并入本文。
64.在某些实施方式中,去除不需要的遗传物质和分离扎结探针可以使用结合剂进行,该结合剂特异性地结合到与连接探针和/或延伸探针结合的部分并因此存在于扎结探针中。例如,可以将延伸探针的5'端与生物素结合,并且可以使用扎结探针与链霉亲和素的特异性结合来分离扎结探针。类似地,连接探针的3'端可以与生物素结合,并且可以使用扎结探针与链霉亲和素的特异性结合来分离扎结探针。
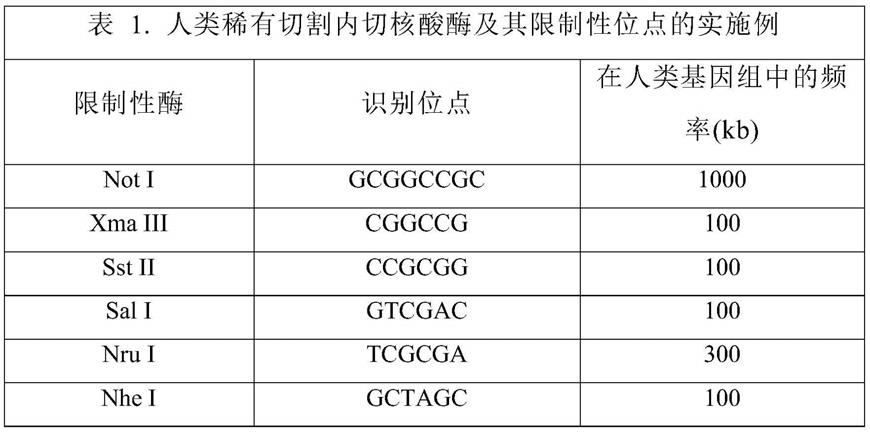
65.可结合至5'或3'端或在连接探针和/或延伸探针内的额外部分以及可用于分离扎结探针的相应结合剂是本领域已知的,并且此类实施方式在本发明的范围内。
66.在某些实施方式中,连接步骤的结束和不需要的材料的任选去除产生扎结探针,其从5'端到3'端包含第一引物结合序列、第一靶结合序列、扩增的靶基因组区域、第二靶结合序列和第二引物结合序列。扎结探针的形成和可选的纯化意味着靶基因组区域的捕获。
67.可以处理扎结探针以制备用于进一步分析的扎结探针。这种处理设计用于三个主要目的,例如通过pcr将扎结探针扩增至可检测水平;合并样品特异性标识符(在本领域中也称为索引、条形码、邮政编码、接头等),以及将某些序列合并到扎结探针中,这些序列有助于扎结探针的测序,从而有助于靶基因组区域的测序。
68.因此,在一些实施方式中,包含以延长的延伸探针形式捕获的靶基因组区域的扎
结探针进行扩增,以产生扎结探针的拷贝。这种扩增可以包括在pcr中使用扩增引物对产生双链形式的扎结探针的拷贝。扩增引物对可以进行设计,使得所得的双链扎结探针,除了靶基因组区域和第一和第二引物结合序列之外,还包括以下中的一个或多个:第一测序引物结合序列、第一标识符序列、第二测序引物结合序列和第二标识符序列。
69.在某些实施方式中,扩增引物对包括:
70.i)延伸探针扩增引物,其从5'到3'端包括以下一个或多个:第一测序引物结合序列、第一标识符序列和第一引物序列,和
71.ii)连接探针扩增引物,其从5'到3'端包括以下一个或多个:第二测序引物结合序列、第二标识符序列和第二引物序列。
72.在优选的实施方式中,扩增引物对包括:
73.i)延伸探针扩增引物,其从5'到3'端包括:第一测序引物结合序列、第一标识符序列和第一引物序列,和
74.ii)连接探针扩增引物,其从5'到3'端包括:第二测序引物结合序列、第二标识符序列和第二引物序列。
75.在该步骤中,利用包含延伸探针扩增引物和连接探针扩增引物的扩增引物对,使用pcr来扩增扎结探针。连接探针扩增引物结合到扎结探针的3'端,即朝向扎结探针的连接探针侧。延伸探针扩增引物与扎结探针5'端的互补物结合,即朝向扎结探针的延伸探针侧。
76.延伸探针扩增引物从5'到3'端包括第一测序引物结合序列、任选地第一标识符序列和第一引物序列。第一引物序列与朝向扎结探针5'端存在的第一引物结合序列的互补物杂交。第一引物结合序列作为延伸探针的一部分被引入扎结探针中。
77.连接探针扩增引物从5'到3'端包括第二测序引物结合序列、任选地第二标识符序列和第二引物序列。第二个引物序列与朝向扎结探针3'端存在的第二引物结合序列杂交。第二引物结合序列作为连接探针的一部分被引入扎结探针。
78.在某些实施方式中,扩增引物对的一个或两个引物包含可以促进在扩增步骤结束时产生的双链靶基因组区域的下游测序的附加序列。可促进测序的附加序列可包含例如流动细胞结合和测序引物结合所需的序列的至少一部分以在illumina
tm
平台上启动测序(例如双端或单读测序),基于发夹接头的测序(例如pacbio测序)所需的发夹接头的至少一部分,或正确引导分子通过基于纳米孔技术的测序仪所需的序列的至少一部分。当所得分子仅包含测序所需的序列的一部分时,其余部分可以通过本领域已知的任何其他方式(例如接头连接)引入。
79.扎结探针和扩增引物对的混合物进行pcr。
80.除了扎结探针和扩增引物对之外,pcr反应混合物可以包含dna聚合酶和其他用于pcr的试剂,例如脱氧核糖核苷酸(dntp)、金属离子(例如,mg
2+
和mn
2+
)和缓冲液。可用于pcr反应的其他试剂是本领域普通技术人员众所周知的,并且此类实施方式在本发明的范围内。
81.通常,pcr包括25到40个循环,每个循环包括在不同温度下的变性、退火和延长的步骤。最终延长步骤可以在pcr的最后一个循环结束时进行。设计pcr的各个方面,包括循环次数和循环内各个步骤的持续时间和温度,对本领域普通技术人员来说是显而易见的,并且此类实施方式在本发明的范围内。
82.当连接探针扩增引物与扎结探针杂交时,会产生图1步骤4中提供的结构。因此,在pcr的初始循环期间,扎结探针的互补拷贝利用扩增引物的所有成分产生。在pcr的第二个循环中,延伸探针扩增引物与扎结探针的互补拷贝结合。
83.在pcr结束时,会产生包含靶基因组区域的双链形式的扎结探针的多个拷贝,其适用于进一步分析,例如检测或测序。这种双链dna的一个实施例在图1的步骤5中提供。该双链dna从一端到另一端包含对应于以下一个或多个的序列:第一测序引物结合序列、第一标识符序列、第一引物序列、第一靶序列、靶基因组区域、第二靶序列、第二引物序列、第二标识符、第二测序引物结合序列,以及可以促进含有靶基因组区域的双链dna的测序的任何附加序列。
84.可以使用本领域已知的技术检测扩增的靶基因组区域,例如使用与靶基因组区域内的序列互补的标记探针。例如,可以基于反应的浊度、荧光检测或标记的分子信标来检测扩增的靶基因组区域。
85.术语“标记”是指可通过光谱、光化学、生物化学、免疫化学、化学或其他物理方式检测的分子。例如,有用的标记包括荧光染料(荧光团)、荧光猝灭剂、发光剂、电子致密试剂、生物素、地高辛、
32
p和其他同位素或其他可以(例如通过结合到寡核苷酸而)被检测到的分子。该术语包括标记试剂的组合,例如荧光团的组合,每个荧光团例如在特定波长或波长组合提供独特的可检测特征。
86.示例性荧光团包括但不限于alexa染料(例如,alexa350、alexa430、alexa488等)、amca、bodipy630/650、bodipy650/665、bodipy-fl、bodipy-r6g、bodipytmr、bodipy-trx、瀑布蓝、cy2、cy3、cy5、cy5.5、cy7、cy7.5、dylight染料(dylight405、dylight488、dylight549、dylight550、dylight649、dylight680、dylight750、dylight800)、6-fam、荧光素、fitc、hex、6-joe、俄勒冈绿488、俄勒冈绿500、俄勒冈绿514、太平洋蓝、reg、罗丹明绿、罗丹明红、rox、r-藻红蛋白(r-pe)、星光蓝染料(例如,星光蓝520、星光蓝700)、tamra、tet、四甲基罗丹明、德克萨斯红和tritc。
87.还可以使用本领域已知的技术对扩增的靶基因组区域进行测序,例如纳米孔测序(oxford nanopore technologies
tm
)、可逆染料终止子测序(illumina
tm
)和单分子实时(smrt)测序(pacbio
tm
)。多种测序仪器可用于测序,例如使用便携式nanoporeminion
tm
或台式机、nanoporepromethion
tm
、pacbiosequel
tm
或illuminahiseq
tm
。测序步骤还可用于多个靶的多重检测和/或多态性检测。优选地,扩增的靶基因组区域的测序在高通量测序仪(例如illumina、pacbio或nanopore装置)上进行,。
88.本领域普通技术人员可以认识到,根据测定的具体方面,例如用于对靶基因组区域进行测序的技术或靶基因组区域的长度,人们可能不需要在扩增步骤期间引入如上所述的所有序列。例如,可以设计扩增引物对,其中一个或两个标识符序列不存在。如果仅研究一个靶基因组区域,则可能不需要标识符序列。此外,如果靶基因组区域较短,例如小于约500bp,则可能不需要两个标识符序列。
89.此外,可以设计扩增引物对,其中一个或两个测序引物结合序列不存在。例如,如果靶基因组区域较短,例如小于约500bp,或者测序(例如pacbio)需要单个测序引物,那么对于测序目的可能仅一个测序引物结合序列就已足够。在某些情况下,连接和延伸探针可能已经包含测序所需的序列(例如测序引物结合序列)的至少一部分。也可以通过扩增引物
对的一个或两个引物引入可以促进含有靶基因组区域的双链dna的测序的任何附加序列。此外,测序引物结合序列可以不存在,而是可以引入促进双链扩增连接探针的进一步加工和后续测序的序列。这样的序列包括限制性酶位点,特别是稀有切割限制性酶位点。
90.稀有切割限制性内切核酸酶的非限制性实施例描述于pct公开wo2009/079488中,其通过引用整体并入本文,特别是表1。
91.如本文所用,“稀有切割限制性内切核酸酶”是其限制性位点很少出现在遗传物质中的内切核酸酶。例如,对于人类基因组,稀有切割限制性内切核酸酶是其限制性位点平均每50-100kb、优选地每100-200kb或更优选地每200-400kb、甚至更优选地每400-600kb出现的内切核酸酶。下表1给出了人类基因组的稀有切割限制性内切核酸酶及其限制性位点的实施例:
[0092][0093]
额外的稀有切割内切核酸酶描述于例如“nucleic acids and molecular biology,pingoud(编者),springer;第一版(2004)”的restriction endonucleases中。许多稀有切割内切核酸酶也可商购获得,例如归巢类内切核酸酶,例如来自newenglandbiolabs(beverly,ma)。稀有切割内切核酸酶的更进一步的实施例是本领域已知的,并且这样的实施方式在本发明的范围内。
[0094]
可以用相应的限制性酶处理稀有切割限制性酶位点,以产生具有粘性末端的双链扩增扎结探针。扩增的靶基因组区域的切割拷贝的粘性末端可用于将靶基因组区域与发夹接头结合。例如,包含与通过扩增引物对引入扩增的靶基因组区域的限制性位点互补的突出端的发夹接头可以与用限制性酶处理的扩增靶基因组区域的拷贝混合,以产生双链靶基因组区域,其包含两侧是其它序列中的发夹接头的靶基因组区域。
[0095]
对应于iis型限制性酶的额外限制性内切核酸酶位点可用于产生双链扩增扎结探针,包括用于尾交换。在优选的实施方式中,使用bsai和mlyi限制性内切核酸酶位点。mlyi提供了一个平端,而bsai提供了一个突出端。下表2中提供了iis型限制性内切核酸酶的实施例、它们的识别位点和使用相应酶消化后的核苷酸突出端数量(如果有):
[0096]
[0097]
[0098][0099]
在某些实施方式中,多个靶基因组区域被捕获并任选地进一步分析,例如检测或测序。对于多个靶基因组区域,使用多对延伸和连接探针。每对延伸和连接探针包含独特的第一和第二靶结合序列,这取决于靶基因组区域两侧的序列。然而,多对延伸和连接探针中的每一对可以具有相同的第一引物结合序列和相同的第二引物结合序列。
[0100]
因此,本文公开的材料和方法的某些实施方式提供从遗传物质捕获多个靶基因组区域。该方法包括以下步骤:
[0101]
a)将多对探针与多对靶序列杂交,其中每对靶序列位于来自多个靶基因组区域的一个靶基因组区域的两侧,并且其中每对探针包含一个延伸探针和一个连接探针,并且对于每对探针:
[0102]
i)延伸探针包含朝向3'端的第一靶结合序列和朝向5'端的第一引物结合序列,和
[0103]
ii)连接探针包含朝向5'端的第二靶结合序列和朝向3'端的第二引物结合序列,其中第一靶结合序列和第二靶结合序列分别与位于靶基因组区域的两侧的第一靶序列和第二靶序列结合;
[0104]
b)将延伸探针的3'端延长,直到扩增后的延伸探针的3'端与相应连接探针的5'端相邻;
[0105]
c)将扩增后的延伸探针的3'端与对应的连接探针的5'端连接以产生多个扎结探针,每个扎结探针包含从5'端到3'端的第一引物结合序列、第一靶结合序列、扩增的靶基因组区域、第二靶结合序列和第二引物结合序列。
[0106]
在某些实施方式中,可以除去扎结探针以外的成分,包括一种或多种未结合的探针、未扎结的延长产物和目标遗传物质。
[0107]
上述捕获靶基因组区域的方面,例如设计延伸和连接探针、靶基因组区域的长度、第一和第二引物结合序列,也适用于捕获多个靶基因组区域的当前方法。
[0108]
在某些实施方式中,本文公开的方法包括使用扩增引物对在pcr中扩增多个扎结探针以产生多个双链扎结探针,所述双链扎结探针进一步包含以下一个或多个:第一测序引物结合序列、第一标识符序列、第二测序引物结合序列和第二标识符序列,其中扩增引物对包括:
[0109]
i)延伸探针扩增引物,其从5'到3'端包括以下一个或多个:第一测序引物结合序列、第一标识符序列和第一引物序列,和
[0110]
ii)连接探针扩增引物,其从5'到3'端包括以下一个或多个:第二测序引物结合序列、第二标识符序列和第二引物序列。
[0111]
优选地,扩增引物对包括:
[0112]
i)延伸探针扩增引物,其从5'到3'端包括:第一测序引物结合序列、第一标识符序列和第一引物序列,和
[0113]
ii)连接探针扩增引物,其从5'到3'端包括:第二测序引物结合序列、第二标识符序列和第二引物序列。
[0114]
在某些实施方式中,扩增引物对的一个或两个引物包含可以促进在扩增步骤结束时产生的双链靶基因组区域的下游测序的附加序列。可促进测序的附加序列可包含:例如,流动细胞结合和测序引物结合所需的序列的至少一部分以在illumina
tm
平台上启动测序(例如双端或单读测序),基于发夹接头的测序(例如pacbio测序)所需的发夹接头的至少一部分,或正确引导分子通过基于纳米孔技术的测序仪所需的序列的至少一部分。当所得分子仅包含测序所需序列的一部分时,其余部分可以通过本领域已知的任何其他方式(例如接头连接)引入。
[0115]
为了从遗传物质中捕获多个靶基因组区域,探针对被设计为包含相同的第一和第二引物结合序列。因此,仅一个扩增引物对可用于从一个样品扩增多个捕获的靶基因组区域。此外,如果进行了后续测序反应,可以使用相同的第一和第二测序引物对多个捕获的靶基因组区域进行测序。因此,来自扩增引物对的一个引物包含以下一个或多个:第一测序引物结合序列、第一标识符序列和第一引物序列,而来自扩增引物对的另一个引物包含以下一个或多个:第二测序引物结合序列、第二标识符序列和第二引物序列。第一和第二标识符序列可以彼此相同,并且第一和第二引物序列可以彼此相同。
[0116]
因此,在捕获多个靶基因组区域的方法的扩增步骤结束时,产生多个扩增的基因组区域的拷贝,每个拷贝包含:第一测序引物结合序列、第一标识符序列、第一引物序列、多个靶基因组区域之一、第二引物序列、第二标识符序列和第二测序引物序列。
[0117]
在某些实施方式中,多个靶基因组区域被进一步分析,例如,检测或测序。可以使用本领域已知的技术,例如使用与靶基因组区域内的序列互补的多个标记探针,检测扩增的靶基因组区域。例如,可以基于反应的浊度、荧光检测或标记的分子信标来检测扩增的靶基因组区域。检测靶基因组区域的上述方面也适用于检测多个基因组区域。
[0118]
也可以使用本领域已知的技术对多个扩增的靶基因组区域进行测序。检测靶基因组区域的上述方面也适用于检测多个基因组区域。
[0119]
特别地,在某些实施方式中,来自多个样品的多个靶基因组区域被合并和测序。在此类实施方式中,获得对应于来自多个样品的多个靶基因组区域的多个序列读数。对于特定读数,唯一的第一和/或第二标识符序列用于将该读数分配给相应的样品,并将读数中捕获的靶基因组区域的序列与已知数据库进行比较,以将序列分配给样品中的靶基因组区域。因此,虽然仅可使用一或两个测序引物对一个反应混合物中的许多靶基因组区域进行测序,但每个测序读数都可以系统地且准确地归因于适当的源样品和适当的靶基因组区域。
[0120]
在某些实施方式中,使用包含两个序列标识符的独特组合的扩增引物对扩增来自多个样品的一个样品中的多个靶基因组区域。因此,多个样品中没有两个样品具有相同的第一标识符和第二标识符组合。例如,十二个唯一的第一标识符和八个唯一的第二标识符可用于产生第一和第二标识符的九十六个唯一组合。因此,使用仅20个标识符的不同组合,就可以唯一地标识96个样品。
[0121]
在这样的实施方式中,对于特定读数,唯一的第一标识符序列和第二标识符序列用于将读数分配给相应的样品,并将读数中捕获的靶基因组区域的序列与已知数据库进行比较,以分配序列到样品中的靶基因组区域。因此,虽然可使用仅一或两个测序引物对一个反应混合物中的许多靶基因组区域进行测序,但每个测序读数都可以系统地且准确地归因
于适当的源样品和适当的靶基因组区域。
[0122]
与检测或测序单个靶基因组区域类似,本领域普通技术人员可以认识到,取决于如何设计扩增引物对,扩增引物对中的一些序列可能不存在。例如,可以只存在一个标识符序列或可以只存在一个测序引物结合序列,特别是当被分析的靶基因组区域很短、例如小于约500bp时,或者测序(例如pacbio)需要单个测序引物时。在某些情况下,连接和延伸探针可能已经包含测序所需的序列(例如测序引物结合序列)的至少一部分。也可以通过扩增引物对的一个或两个引物引入可以促进含有靶基因组区域的双链dna的测序的任何附加序列。此外,两个测序引物结合序列可以不存在,而是可以引入促进双链扩增的靶基因组区域的进一步加工和后续测序的序列。这样的序列包括限制性酶位点,特别是稀有切割限制性酶位点。上文讨论的稀有切割限制性酶也可用于本发明的这些实施方式中。
[0123]
还设想了用于实施本文公开的方法的试剂盒。某些此类试剂盒可包含设计用于捕获一个或多个靶基因组区域的特定延伸探针和连接探针、用于扩增一个或多个捕获的靶基因组区域的连接探针扩增引物、延伸探针扩增引物、dna连接酶、聚合酶和其他用于pcr的试剂、测序试剂、设计用于处理从测定中获得的测序数据的计算机软件程序以及可选的提供执行测定的指令的材料。
[0124]
在某些实施方式中,可以针对一个或多个特定靶基因组区域定制试剂盒。例如,用户可以提供一个或多个靶基因组区域的序列,并且可以生产试剂盒以执行本文公开的用于分析一个或多个靶序列的测定。
[0125]
在本文公开的方法中使用的延伸和连接探针的合成通常是昂贵的,特别是如果探针包含修饰,例如5'磷酸酯和/或修饰的寡核苷酸。例如,商业供应商用于合成此类探针的常规方法涉及亚磷酰胺方法。这种方法包括一次合成和纯化一个寡核苷酸,最终产生一批单链寡核苷酸。
[0126]
本文公开的分析靶基因组区域的方法需要每个靶区域的两个寡核苷酸,因为连接和延伸探针结合靶基因组区域两侧的序列。传统技术使用等位基因特异性寡核苷酸,因此每个双等位基因靶基因组区域需要三个寡核苷酸。本文公开的方法提供了对常规方法的改进,因为这些方法每个靶基因组区域仅需要两个寡核苷酸。
[0127]
即使与每个靶基因组区域三个寡核苷酸相比,每个靶基因组区域两个寡核苷酸的合成成本降低,合成一对连接和延伸探针的成本也需要进一步降低。考虑到所要求保护的方法可用于同时分析多个靶基因组区域,合成一对连接和延伸探针的成本的降低以指数方式反映在设计用于分析数千个靶基因组区域的测定的总成本节约中。
[0128]
为此,本发明的某些实施方式提供了用于产生双链形式的连接和/或延伸探针的方法,以用于本文公开的方法中。这些方法是可扩展的,并且通常将合成寡核苷酸的成本降低至少10倍,甚至可能降低100到1,000倍。
[0129]
本发明的某些实施方式提供了一种从单链寡核苷酸产生双链探针的方法。该方法设计用于生产双链探针,该探针双链靶向染色体的两条链并分别构成相对于每条链的延伸和连接探针。这种单链寡核苷酸在本文中被称为“单链预探针”。为了允许添加修饰和包含测序所需的序列(例如测序引物结合序列)的至少一部分,可以产生两组或更多组探针。作为在illumina平台上测序的实施例,构建了两组探针,本文定义为上游和下游探针,它们分别包含对应于i5和i7illumina接头序列的至少一部分的序列。在某些实施方式中,上游双
链探针杂交靶区域的左侧,而下游双链探针靶向靶区域的右侧。双链连接和延伸上游探针的生产方法包括以下步骤:
[0130]
a)提供单链或双链预探针,其从5'端到3'端包含:第一尾部、第一限制性酶的第一限制性位点、靶结合序列、第二限制性酶的第二限制性位点、以及第二尾部,其中双链预探针任选地在pcr中使用合适的引物复制单链预探针而产生,
[0131]
b)任选地,进行尾交换反应以用临时的第一或第二尾部代替经遗传修饰以包含新的所需序列的至少一部分的永久尾部,包括:
[0132]
i)用第一限制性酶消化双链上游前(pre-upstream)探针,以去除第一上游前尾部或它的一部分,以产生突出端,和
[0133]
ii)将用第一限制性酶消化的双链上游前探针连接到永久尾部,永久尾部分子包含遗传修饰,并且上游永久尾部的至少一部分包含与消化的双链上游前探针的突出端互补的突出端,
[0134]
c)任选地,纯化与永久尾部连结的双链上游前探针,
[0135]
d)通过用第二限制性酶消化含有第一尾部或替换第一尾部的永久尾部的双链上游前探针以去除第二尾部并在第一靶结合序列内产生粘性末端或者优选地平端,来产生双链上游探针,和
[0136]
e)任选地,纯化双链上游探针。在一些实施方式中,双链探针可以通过本领域已知的方法转化为单链探针,例如双链探针的变性或通过选择性降解一条链。
[0137]
在优选的实施方式中,第一限制性酶是消化双链dna以产生远离其识别位点的粘性末端的iis型,并且第二限制性酶是消化远离其识别位点的双链dna并且在dna中产生平端切口的另一种iis型。在甚至优选的实施方式中,第一限制性酶是bsai并且第二限制性酶是mlyi。在某些实施方式中,突出端可以是至少1、2、3、4、5或更多个核苷酸。在优选的实施方式中,突出端是1-5个核苷酸,更优选1-3个核苷酸,最优选1-2个核苷酸。
[0138]
在某些实施方式中,条形码置于第一限制性位点和靶结合序列和/或靶结合序列和第二限制性位点之间。
[0139]
在某些实施方式中,探针构建可以直接从双链上游前探针开始,因此跳过将单链分子转化为双链分子的第一pcr步骤或仅进行pcr以扩增上游前探针的数量。
[0140]
用第一限制性酶消化的步骤和用第二限制性酶消化的步骤的顺序可以互换或同时发生。特别地,可以首先执行产生优选平端的利用第二限制性酶的消化,然后用第一限制性酶消化产生突出端,并与第一交换接头连接。无论消化顺序如何,在消化和连接结束时都会产生相同的双链上游探针。
[0141]
如图3所示,可以在没有尾部交换步骤的情况下构建探针。在没有尾部交换步骤的情况下,进行限制性酶消化以去除不需要的尾部并激活用于杂交的探针。可以使用至少1、2、3、4或更多种限制性酶进行单一消化反应。或者,可以连续地进行消化反应,其中在添加后续限制性酶之前去除或灭活一种限制性酶。消化反应产生的dna可能有一个平端。可以使用本领域技术人员熟知的方案,例如使用dna聚合酶i的klenow片段,去除反应产生的任何单链突出端或填充凹陷链,以形成平端。
[0142]
如图4所示,上游探针以双链形式生产。在5'端具有修饰的上链对应于本公开中较早讨论的延伸探针。在3'端具有修饰的另一条链对应于本公开中较早讨论的连接探针。因
此,在根据本文公开的方法产生的双链上游探针中,一条链是适用于分析靶基因组区域的一条链的延伸探针,而另一条链是适用于分析靶基因组区域的另一条链的连接探针。
[0143]
本发明的进一步实施方式提供了一种从设计为靶向靶区域右侧的单链寡核苷酸产生双链下游探针的方法。这种单链寡核苷酸在本文中被称为“单链下游前探针”。双链下游探针的制备方法包括以下步骤:
[0144]
a)提供单链或双链预探针,其从5'端到3'端包含:第一尾部、第一限制性酶的第一限制性位点、靶结合序列、第二限制性酶的第二限制性位点和第二尾部,其中双链预探针任选地在pcr中使用合适的引物复制单链预探针而产生,
[0145]
b)任选地,进行尾交换反应以用临时的第一或第二尾部代替经遗传修饰以包含新的所需序列的至少一部分的永久尾部,包括:
[0146]
i)用第二限制性酶消化双链下游前探针以去除第二下游前尾部或它的一部分,以产生突出端,和
[0147]
ii)将用第二限制性酶消化的双链下游前探针连接到永久尾部上,永久尾部分子包含遗传修饰,并且下游永久尾部的至少一部分包括与消化的双链下游前探针的突出端互补的突出端,
[0148]
c)任选地,纯化与永久尾部连接的双链下游前探针,
[0149]
d)用第一限制性酶消化具有第二尾部或替代第二尾部的永久尾部的双链下游前探针,以去除第一下游尾部并在第二靶结合序列内产生粘性末端或者优选地平端,来产生双链下游探针,和
[0150]
e)可选地,纯化双链下游探针。在一些实施方式中,双链探针可以通过本领域已知的方法转化为单链探针,例如双链探针的变性或通过选择性降解一条链。
[0151]
在优选的实施方式中,第二限制性酶是消化双链dna以产生远离其识别位点的粘性末端的iis型,并且第一限制性酶是消化远离其识别位点的双链dna并且优选地在dna中产生平端切口的另一种iis型。在甚至优选的实施方式中,第二限制性酶是bsai并且第一限制性酶是mlyi。
[0152]
在某些实施方式中,条形码置于第一限制性位点和靶结合序列和/或靶结合序列和第二限制性位点之间。
[0153]
在某些实施方式中,探针构建可以直接从双链下游前探针开始,因此跳过将单链分子转化为双链分子的第一pcr步骤或简单地进行pcr以扩增上游前探针的数量。
[0154]
用第二限制性酶消化的步骤和用第一限制性酶消化的步骤的顺序可以互换或同时发生。特别地,可以首先执行优选产生平端的第一限制性酶消化,然后用第二限制性酶消化产生突出端并与第二外切核酸酶保护的接头连接。在某些实施方式中,突出端可以是至少1、2、3、4、5或更多个核苷酸。在优选的实施方式中,突出端是1-5个核苷酸,更优选1-3个核苷酸,最优选1-2个核苷酸。无论消化顺序如何,在消化和连接结束时都会产生相同的双链连接探针。
[0155]
如图4所示,连接探针以双链形式生产。在5'端具有修饰的上链对应于本公开中较早讨论的延伸探针。在3'端具有修饰的另一条链对应于本公开中较早讨论的连接探针。因此,在根据本文公开的方法产生的双链下游探针中,一条链是适合于分析靶基因组区域的一条链的连接探针,而另一条链是适合于分析靶基因组区域的另一条链的延伸探针。
[0156]
图5描述了生产双链上游和下游探针的一般方案。本领域普通技术人员将理解,在根据上述方法产生的双链上游探针中,在5'端包含修饰的链可用作延伸探针,而在3'端包含修饰的链可用作连接探针。相反,在根据上述方法生产的双链下游探针中,在3'端包含修饰的链可用作连接探针,在5'端包含外切核酸酶保护的链可用作延伸探针。因此,本发明的一个实施方式提供了一种生产在一端在双链上具有遗传修饰的双链寡核苷酸探针的方法,该方法包括:
[0157]
a)提供单链或双链预探针,其从5'端到3'端包括:包含第一限制性酶的第一限制性位点的第一尾部、靶结合序列、包含第二限制性酶的第二限制性位点的第二尾部,其中双链预探针任选地在pcr中使用合适的引物复制单链预探针而产生,
[0158]
b)任选地,进行尾部交换反应以用临时的第一尾部或第二尾部代替经遗传修饰以包含新的所需序列的至少一部分的永久尾部,包括:
[0159]
i)用一种限制性酶消化双链预探针以去除一个尾部或其一部分,以产生突出端,并且
[0160]
ii)将用限制性酶消化的双链探针连接到永久尾部,永久尾部分子包含遗传修饰,永久尾部的至少一部分包括与消化的双链探针的突出端互补的突出端,
[0161]
c)任选地,纯化连接到永久尾部的双链预探针,
[0162]
d)用一种限制性酶消化具有一个尾部或替代该一个尾部连接的永久尾部的双链预探针,以去除一个尾部,或者在一条尾部已被交换的情况下去除另一个尾部,并在靶结合序列内产生粘性末端或优选平端,从而产生双链探针,并且
[0163]
e)任选地,纯化双链探针。在一些实施方式中,双链探针可以通过本领域已知的方法转化为单链探针,例如双链探针的变性或通过选择性降解一条链。
[0164]
在优选的实施方式中,一种限制性酶是消化双链dna以产生远离其识别位点的粘性末端的iis型,并且另一种限制性酶是消化远离其识别位点的双链dna并且优选地在dna中产生平端切口的另一种iis型。在甚至优选的实施方式中,一种限制性酶是bsai,另一种限制性酶是mlyi。
[0165]
在某些实施方式中,条形码置于第一限制性位点和靶结合序列和/或靶结合序列和第二限制性位点之间。
[0166]
在某些实施方式中,探针构建可以直接从双链预探针开始,因此跳过将单链分子转化为双链分子的第一pcr步骤。
[0167]
用第二限制性酶消化的步骤和用第一限制性酶消化的步骤的顺序可以互换或同时发生。特别地,可以首先执行优选产生平端的限制性酶消化,然后用限制性酶消化产生突出端并与交换的接头连接。在某些实施方式中,突出端可以是至少1、2、3、4、5或更多个核苷酸。在优选的实施方式中,突出端是1-5个核苷酸,更优选1-3个核苷酸,最优选1-2个核苷酸。无论消化顺序如何,在消化和连接结束时都会产生相同的双链连接探针。
[0168]
双链探针可用于捕获和分析双链基因组的靶基因组区域的两条链。因此,本发明的某些实施方式提供了一种从双链目标遗传物质中捕获靶基因组区域的方法。该方法包括以下步骤:
[0169]
a)提供一对双链探针,其中每个双链探针的每条链分别对应连接和延伸探针,
[0170]
其中目标上游的双链探针包含:
[0171]
i)第一延伸探针,包括朝向3'端的第一靶结合序列和朝向5'端的第一引物结合序列,和
[0172]
ii)第二连接探针,包括朝向5'端的第一靶结合序列和朝向3'端的第一引物结合序列,
[0173]
并且其中目标下游的双链探针包含:
[0174]
i)第一连接探针,包括朝向5'端的第二靶结合序列和朝向3'端的第二引物结合序列,和
[0175]
ii)第二延伸探针,包括朝向3'端的第二靶结合序列和朝向5'端的第二引物结合序列;
[0176]
b)将双链靶基因组区域与双链延伸探针和双链连接探针接触,在允许以下情况的条件下进行接触:
[0177]
i)双链上游探针和双链下游探针变性为第一延伸探针、第二连接探针、第一连接探针和第二延伸探针,和
[0178]
ii)第一延伸探针和第一连接探针与靶基因组区域中的第一条dna链杂交,以及第二延伸探针和第二连接探针与靶基因组区域中的第二条dna链杂交,
[0179]
c)扩增第一延伸探针的3'端,直到扩增的第一延伸探针的3'端与第一连接探针的5'端相邻,并扩增第二个延伸探针的3'端,直到扩增的第二延伸探针的3'端与第二连接探针的5'端相邻;和
[0180]
d)通过以下方式从双链目标遗传物质中捕获靶基因组区域:
[0181]
i)将扩增的第一延伸探针的3'端与第一连接探针的5'端连接以产生第一扎结探针,第一扎结探针从5'端到3'端包含第一引物结合序列、第一靶结合序列、扩增的靶基因组区域、第二靶结合序列和第二引物结合序列,以及
[0182]
ii)将扩增的第二延伸探针的3'端与第二连接探针的5'端连接以产生第二扎结探针,第二扎结探针从5'端到3'端包含第二引物结合序列、第二靶结合序列、扩增的靶基因组区域、第一靶结合序列和第一引物结合序列。
[0183]
本领域普通技术人员将理解,在一对双链上游探针和双链下游探针中,上游探针和下游探针中的靶结合序列被设计为使得它们位于靶基因组区域的两侧。
[0184]
此外,上述合成双链上游和下游探针的方法可用于生产本文公开的方法中使用的双链探针,用于捕获靶基因组区域的两条链。因此,在某些实施方式中,上游探针在第一延伸探针的5'端包含外切核酸酶保护,在第二连接探针的3'端包含外切核酸酶保护。类似地,下游探针在第一连接探针的3'端包含外切核酸酶保护,在第二延伸探针的5'端包含外切核酸酶保护。
[0185]
延伸和/或连接探针末端的外切核酸酶保护可以包括以下一种或多种:在核苷酸之间引入硫代磷酸酯键,合并两个或多个亚磷酰胺和单硫代磷酸酯和/或二硫代磷酸酯键,通过甲缩醛/缩酮键代替相邻核苷酸之间的一个或多个磷酸二酯键,通过磷酰基或乙酰基封闭3'末端羟基,引入3'末端氨基磷酸酯,引入肽核酸(pna)或锁核酸(lna),引入一个或多个硫代磷酸酯基团,或引入寡核苷酸骨架中的2-o-甲基核糖基团。
[0186]
在产生第一和第二扎结探针后,可以将它们从反应混合物中分离。这种分离可以包括通过用一种或多种具有5'-3'外切核酸酶活性和3'-5'外切核酸酶活性的外切核酸酶
处理反应混合物来消化反应混合物中不需要的部分,例如未结合的探针或靶基因组dna。因为第一和第二扎结探针在5'和3'端都具有保护,可以使用提供5'-3'外切核酸酶活性和3'-5'外切核酸酶活性的外切核酸酶或外切核酸酶组合.
[0187]
靶基因组区域可以在约10bp和约100bp之间、在约100bp和约300bp之间、在约300bp和约1000bp之间或在约1000bp和约20000bp之间。
[0188]
一旦分离,可以使用特定引物对扩增第一和第二扎结探针。因此,分析捕获的靶基因组区域的进一步步骤包括:在聚合酶链式反应(pcr)中使用扩增引物对扩增第一和/或第二扎结探针,以产生双链形式的第一和/或第二扎结探针的拷贝,其中第一扎结探针扩增引物对包括:
[0189]
i)第一延伸探针扩增引物,其从5'到3'端包括以下的一个或多个:第一测序引物结合序列、第一标识符序列和第一引物序列,和
[0190]
ii)第一连接探针扩增引物,其从5'到3'端包括以下的一个或多个:第二测序引物结合序列、第二标识符序列和第二引物序列;且
[0191]
其中第二连接探针扩增引物对包括:
[0192]
i)第二延伸探针扩增引物,其从5'到3'端包括以下的一个或多个:第三测序引物结合序列、第三标识符序列和第二引物序列,和
[0193]
ii)第二连接探针扩增引物,其从5'到3'端包括以下的一个或多个:第四测序引物结合序列、第四标识符序列和第一引物序列。
[0194]
本领域技术人员可以为第一和第二引物结合序列设计合适的序列,它们可以相同或不同。此外,第一、第二、第三和第四测序引物可以具有相同或不同的序列。优选地,第一、第二、第三和第四标识符序列具有不同的序列。
[0195]
一旦被扩增,双链扎结探针就可以如本公开前面关于捕获靶基因组区域的单链的方法所讨论的进行测序。
[0196]
与单链探针的设计类似,靶序列位于靶基因组区域的两侧。此外,第一和第二延伸探针3'端的序列与遗传物质上相应的靶序列杂交,第一和第二连接探针5'端的序列与遗传物质上相应的靶序列杂交。因此,延伸探针和连接探针与相应的靶序列结合,并且这些靶序列位于靶基因组区域的两侧。此外,在某些实施方式中,每个延伸和连接探针与位于靶基因组区域两侧的第一和第二靶序列不相邻地杂交。此外,双链延伸和连接探针上的第一和第二引物结合序列可以相同或不同。
[0197]
本领域普通技术人员将理解,本文中使用的每个双链探针可以被称为“双链上游探针”或“双链下游探针”,因为可以使用每个双链探针的一条链作为连接探针,另一条链可以用作延伸探针。因此,本文中使用的描述为了便于参考,将探针之一称为“双链下游探针”,且将另一个探针称为“双链上游探针”。图4底部提供了“双链下游探针”和“双链上游探针”组合的实施例。
[0198]
类似地,存在于第一延伸探针中的第一靶结合序列与存在于第二连接探针中的第一靶结合序列反向互补。此外,存在于第一连接探针中的第二靶结合序列与存在于第二延伸探针中的第二靶结合序列反向互补。因此,此处所用的对“第一靶结合序列”和“第二靶结合序列”的描述是为了便于参考。这些序列之间的这种关系从图4底部提供的双链延伸探针和双链连接探针的组合中可以明显看出。因此,当产生第一和第二扎结探针时,两个扎结探
针包含反向互补的靶基因组区域的拷贝。因此,靶基因组区域的两条链都被捕获。
[0199]
双链上游探针和双链下游探针中的每一个可包含最少约20至约200个核苷酸。特别地,第一靶结合序列可以至少在约10和约60个核苷酸之间。第一引物结合序列也可以至少在约10至约30个核苷酸之间。类似地,第二靶序列可以至少在约10至约60个核苷酸之间,并且第二引物结合序列可以至少在约10至约30个核苷酸之间。探针对靶结合位点的特异性可由第一和第二靶结合序列的长度控制。特别地,第一和第二靶结合序列的较长长度提供更高的结合特异性,而第一和第二靶结合序列的较短长度提供较低的特异性。本领域普通技术人员可以基于靶基因组区域的序列和特定生物体的可用基因组序列,例如从基因组序列数据库确定第一和第二靶结合序列的合适序列。
[0200]
上面关于单链延伸和连接探针讨论了杂交、延伸、连接、去除不需要的材料、扎结探针的扩增、探针的外切核酸酶保护、样品特异性标识符(在本领域中也称为索引、条形码、邮政编码、接头等)合并以及靶基因组区域的测序的细节。这些细节也适用于本文所用的利用双链探针的方法。
[0201]
在某些实施方式中,多个靶基因组区域被捕获并任选地进一步分析,例如检测或测序。对于多个靶基因组区域,使用多对双链上游和下游探针。每对双链上游和下游探针包含独特的第一和第二靶结合序列,这取决于靶基因组区域两侧的序列。然而,多对上游和下游探针中的每一对可以具有相同的第一引物结合序列和/或相同的第二引物结合序列。
[0202]
因此,本文公开的材料和方法的某些实施方式提供从遗传物质捕获多个靶基因组区域。该方法包括以下步骤:
[0203]
a)将多对双链探针与多对靶序列杂交,其中每对靶序列位于来自多个靶基因组区域的一个靶基因组区域的两侧,并且其中每对探针包含双链上游探针和双链下游探针,每对双链探针包括:
[0204]
一种双链上游探针,其包括:
[0205]
i)第一延伸探针,包括朝向3'端的第一靶结合序列和朝向5'端的第一引物结合序列,和
[0206]
ii)第二连接探针,包括朝向5'端的第一靶结合序列和朝向3'端的第一引物结合序列,
[0207]
和一种双链下游探针,其包括:
[0208]
i)第一连接探针,包括朝向3'端的第二靶结合序列和朝向5'端的第二引物结合序列,和
[0209]
ii)第二延伸探针,包括朝向3'端的第二靶结合序列和朝向5'端的第二引物结合序列;
[0210]
其中第一靶结合序列和第二靶结合序列分别与位于靶基因组区域两侧的第一靶序列和第二靶序列结合;
[0211]
b)扩增第一延伸探针的3'端,直到扩增的第一延伸探针的3'端与第一连接探针的5'端相邻,并扩增第二延伸探针的3'端,直到扩增的第二延伸探针的3'端与第二连接探针的5'端相邻;和
[0212]
c)通过以下方式从双链目标遗传物质中捕获多个靶基因组区域:
[0213]
i)将扩增的第一延伸探针的3'端与第一连接探针的5'端连接以产生多个第一扎
结探针,每个第一扎结探针从5'端到3'端包括第一引物结合序列、第一靶结合序列、扩增的靶基因组区域、第二靶结合序列和第二引物结合序列,和
[0214]
ii)将扩增的第二延伸探针的3'端与第二连接探针的5'端连接以产生多个第二扎结探针,每个第二扎结探针从5'端到3'端包括第二引物结合序列、第二靶结合序列、扩增的靶基因组区域、第一靶结合序列和第一引物结合序列。
[0215]
上述捕获靶基因组区域的方面,例如设计双链上游和下游探针、靶基因组区域的长度、第一和第二引物结合序列,也适用于使用多个双链上游和下游探针捕获多个靶基因组区域的当前方法。
[0216]
在某些实施方式中,多个捕获的靶基因组区域中的每一个可以在包括扎结探针的扩增和测序的方法中测序。以上提供的关于这些步骤的细节也适用于捕获和分析多个双链靶基因组区域的方法。
[0217]
本发明的进一步实施方式还提供包含一对或多对双链探针的试剂盒。每对双链探针包括一个双链上游探针和一个双链下游探针,其中
[0218]
其中双链上游探针包括:
[0219]
i)第一延伸探针,包括朝向3'端的第一靶结合序列和朝向5'端的第一引物结合序列,和
[0220]
ii)第二连接探针,包括朝向5'端的第一靶结合序列和朝向3'端的第一引物结合序列,
[0221]
并且其中双链下游探针包括:
[0222]
i)第一连接探针,包括朝向5'端的第二靶结合序列和朝向3'端的第二引物结合序列,和
[0223]
ii)第二延伸探针,包括朝向3'端的第二靶结合序列和朝向5'端的第二引物结合序列。
[0224]
双链上游探针可以包含在第一延伸探针的5'端的外切核酸酶保护,在第二连接探针的3'端的外切核酸酶保护,并且双链下游探针可以包含在第一连接探针的3'端的外切核酸酶保护和第二延伸探针的5'端的外切核酸酶保护。外切核酸酶保护可以包括以下一种或多种:核苷酸之间的硫代磷酸酯键,两个或多个亚磷酰胺和单硫代磷酸酯和/或二硫代磷酸酯键,通过甲缩醛/缩酮键的相邻核苷酸之间的一个或多个磷酸二酯键,通过磷酰基或乙酰基封闭的3'末端羟基,3'末端氨基磷酸酯、肽核酸(pna)或锁核酸(lna)、一个或多个硫代磷酸酯基团或寡核苷酸骨架中的2-o-甲基核糖基团。
[0225]
下面的实施例说明了实施本发明的程序的一个实施方式。该实施例不应被解释为限制性的。
[0226]
实施例1
–
制备一对双链上游和下游探针
[0227]
产生了两组双链探针,它们将与目标区域周围的dna的两条链杂交。图6说明了使用与靶基因组区域的两条链杂交的双链探针的反应的概览。
[0228]
对于靶基因组区域,设计了一对探针,使其与目标区域的下游和上游杂交。上游探针在本文中被称为左探针或i5探针,而下游探针在本文中被称为右探针或i7探针。与目标杂交的探针区域被称为位点特异性序列(lss),可以基于序列互补性基于靶基因组区域两侧的序列进行设计。为了形成单链预延伸和预连接探针,将四个通用尾部附加到lss各自的
3'和5'端。四个尾部彼此不同,但在所有目标中都是相同的。因此,预探针的序列如下:
[0229]
上游前探针:5'尾部1
–
lss1
–
尾部2:
[0230]
下游前探针:5'尾部3
–
lss2
–
尾部4:
[0231]
其中lss1和lss2具有至少10个碱基,优选地,10至60个碱基。
[0232]
尾部包含任一侧的针对限制性酶的识别序列和pcr引物的结合区。探针构建开始于合成单链形式的预探针寡核苷酸。这些可以单独合成并通过常规方法合并或并行合成。一旦预探针池合成并在溶液中,进行pcr的第一步以产生双链预探针。使用适当的引物进行以两个独立的反应进行pcr,一对引物扩增下游预探针,另一引物对扩增上游预探针:
[0233]
引物名称序列preprobe_pcr_up_1与尾部2杂交preprobe_pcr_up_2与尾部1的补体杂交preprobe_pcr_down_1与尾部4杂交preprobe_pcr_down_2与尾部3的补体杂交
[0234]
处理上游pcr的产物,从而通过优选产生平端的酶消化去除尾部2,并通过酶促反应的组合利用含有修饰的寡核苷酸的接头交换尾部1。类似地,处理下游pcr的产物,从而通过优选产生平端的酶消化去除尾部3,并利用含有修饰寡核苷酸的另一个接头交换尾部4。
[0235]
如图6所示,这些探针是双链的并且彼此完全互补,因此倾向于杂交并保持双链形式。然而,在适当的反应条件(例如变性步骤)下,双链探针被转化为单链形式并与靶基因组区域上的相应序列杂交。双链探针可以通过本领域已知的各种方式(例如变性或通过选择性降解一条链)转化为单链探针。使用双链探针可以捕获靶基因组区域的两条链,并且与使用单链探针的反应相比,实现了更高的效率、均匀性和产量。提高效率的部分原因是捕获目标区域的机会加倍,因为捕获每条链的探针可能具有不同的最佳杂交条件,并且如果一对连接和延伸探针未能杂交,另一对可能仍会结合并捕获目标。提高的一致性的部分原因在于两条链之间的碱基组成和杂交动力学的某些条件在不同靶基因组位点的不同变化内是最佳的。最后,产量增加,部分原因是与使用单链探针的反应相比,捕获的靶基因组区域的数量加倍。
[0236]
在此提及或引用的所有专利、专利申请、临时申请和出版物通过引用整体并入,包括所有附图和表格,只要它们与本说明书的明确教导不矛盾。
[0237]
应当理解,这里描述的实施方式仅用于说明的目的,根据其的各种修改或变化能够被本领域技术人员提出,并且将被包括在本技术的精神和范围内以及所附权利要求的范围内。此外,本文公开的任何发明或其实施方式的任何要素或限制可以与本文公开的任何和/或所有其他要素或限制(单独地或任意组合地)或任何其他发明或其实施方式组合,并且所有此类组合均被考虑在本发明的范围内,但不限于此。
[0238]
参考文献
[0239]
1.campbell,n.r.,harmon,s.a.,and narum,s.r.(2015).genotyping-in-thousands by sequencing(gt-seq):a cost effective snp genotyping method based on custom amplicon sequencing.mol.ecol.resour.15,855
–
867.doi:10.1111/1755-0998.12357.
[0240]
2.gnirke,a.,melnikov,a.,maguire,j.,rogov,p.,leproust,e.m.,brockman,
w.,et al.(2009).solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing.nat biotechnol 27,182
–
189.doi:nbt.1523[pii]10.1038/nbt.1523.
[0241]
3.shen,p.,wang,w.,krishnakumar,s.,palm,c.,chi,a.-k.,enns,g.m.,et al.(2011).high-quality dna sequence capture of 524 disease candidate genes.proc.natl.acad.sci.u.s.a.108,6549
–
54.doi:10.1073/pnas.1018981108.
[0242]
4.united states patent 8,808,991.
[0243]
5.united states patent 8,460,866.
[0244]
6.pct publication wo 2005/118847.
[0245]
7.pct publication wo 2009/079488.
[0246]
8.krishnakumar,s.,zheng,j.,wilhelmy,j.,faham,m.,mindrinos,m.,and davis,r.,pnas(2008)105(27):9296-9301.
[0247]
9.united states patent 8,795,968.
[0248]
10.united states patent application publication number 2008/0026393.
[0249]
11.el-sagheer et al.(2011),pnas;108(28)11338-11343
[0250]
12.u.s.patent 4,656,127.
[0251]
13.shaw et al.,1991,nucleic acids research,19,747-750.
[0252]
14.raney et al.,1998,peptide nucleic acids(nielsen,p.e.,and egholm,m.,eds.)horizon scientific press,wymondham,u.k.
[0253]
15.simeonov et al.,nucl.acids res.2002,vol.30,e31.
[0254]
16.jacobsen et al.,int.biot.lab,feb 2001,18.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1