一种无机纳米粒子动态交联双网络改性天然聚合物材料的制备方法
1.本发明属于材料领域,尤其涉及一种无机纳米粒子动态交联双网络结构改性天然聚合物材料及其制备方法。
背景技术:
2.当前,纺织行业发展迅速且前景良好,目前对于纺织品的开发主要包括天然纤维、再生纤维和合成纤维等。天然聚合物纤维是一种由天然高分子制备的材料,其具有抗菌、可降解、可再生等优良特点,广泛应用于医疗、美容、环保和生物化学等领域。它的出现及发展给研究人员带来了一个新的研究方向。但是,纯天然高分子纤维往往力学性能有所缺陷,不能达到人们所需要的要求,制约了其进一步的应用与发展。因此,近年来出现了许多通过引入其他组分或形成特殊结构对天然高分子纤维改性来提高纤维的力学性能以拓宽其应用领域,例如有机组分、无机组分和双网络结构。
3.近年来,利用天然聚合物为基体制备纤维材料已有专利公开,专利号cn110359110a以再生纤维素纤维为模板,海藻酸钠接枝纤维素浆粕是通过将海藻酸钠和纤维素浆粕在交联剂的作用下进行接枝反应制得的;最终制得的一种海藻酸盐改性再生纤维素纤维,主要由再生纤维素纤维基体以及均匀分散在其中的海藻酸盐组成,海藻酸盐分子链与再生纤维素分子链之间通过交联剂连接。
4.已报道的改性天然聚合物纤维的生产工艺为湿法纺丝或干喷湿法纺丝,既将海藻酸钠接枝纤维素浆粕溶解制得纺丝液后,以水或多价金属盐离子溶液为凝固浴,进行湿法纺丝制得海藻酸盐改性再生纤维素纤维。目前改性天然聚合物纤维普遍采用湿法纺丝法,是通过在天然高分子内部形成离子交联网络,存在改性纤维材料的力学性能差且不均、功能性差等弊端。
技术实现要素:
5.本发明的目的在于提供一种无机纳米粒子动态交联双网络改性天然聚合物材料的制备方法,是有无机纳米粒子结构的、动态交联和增强增韧效果良好的双网络纤维或膜或纤维气凝胶。
6.本发明的目的是这样实现的:一种无机纳米粒子动态交联双网络结构改性天然聚合物材料的方法,是通过“一锅法”来制备,包括如下步骤:
7.在氮气保护的搅拌条件下,将含双键有机单体、无机纳米粒子(vsnp)添加到天然高分子化合物溶液中,再加入引发剂,在30
‑
50℃下自由基聚合12
‑
36h,得到共混溶液;反应结束后,将共混溶液通过湿法纺丝、静电纺丝或离心纺丝的方法进入凝固剂溶液中凝固成型,制成纤维材料,或将混合溶液通过流延法成膜,之后进入凝固剂溶液中凝固成型,制成膜材料。
8.其中,所述无机纳米粒子(vsnp)的制备方法为:含双键硅基无机前驱体在ph=4~
5的乙醇或甲醇和水混合溶液中室温(一般指25℃)下水解20
‑
60min至澄清透明,得到水解产物vsnp;
9.所述天然高分子化合物溶液的质量浓度为1
‑
2%;含双键有机单体占天然高分子化合物质量分数的1%
‑
30%,优选为15%;无机纳米粒子占含双键有机单体质量分数的0.5%
‑
9%,优选为7%;引发剂占含双键有机单体质量分数的1%
‑
10%,优选为2%;凝固剂溶液的质量浓度为1
‑
5%,优选为3%。
10.本发明动态交联双网络结构通过共价键、动态氢键、离子交联和互穿互锁作用协效生成。利用无机纳米粒子的活性双键与含双键有机单体共聚成杂化柔性长链,“一锅法”同步引入天然高分子链,利用天然高分子离子交联网络为刚性第一网络,利用动态氢键伪交联为柔性第二网络,形成动态交联双网络结构,调控分子间作用与流变行为,通过在凝固剂溶液中离子交联凝胶化成形纤维。
11.根据上文技术方案,优选的情况下,所述含双键硅基无机前驱体选自乙烯基三乙氧基硅烷、γ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、乙烯基三甲氧基硅烷及同类前驱体中的至少一种。
12.根据上文技术方案,优选的情况下,含双键有机单体为丙烯酸(aa)、丙烯酰胺、聚乙二醇二丙烯酸酯及它们的衍生物中的至少一种。
13.根据上文技术方案,优选的情况下,所述引发剂选自过硫酸铵(aps)、过硫酸钾、过硫酸钠、过氧化氢、过氧化苯甲酰中的至少一种。
14.根据上文技术方案,优选的情况下,所述天然高分子化合物选自海藻酸钠(sa)、壳聚糖、纤维素、蛋白质中的至少一种。
15.根据上文技术方案,优选的情况下,所述天然高分子化合物的分子量为3
×
106‑5×
106g/mol。
16.根据上文技术方案,优选的情况下,所述凝固成型的时间为1
‑
10min,凝固成型凝固剂为二价或三价金属离子化合物,或二元羧酸、尿素或二元醛。所述二价或三价金属离子化合物可以为cacl2、cuso4、znso4,所述二元羧酸为最重要的是己二酸(aa),还有壬二酸(aza)、癸二酸酐(csa)、间苯二甲酸(ipa)、对苯二甲酸(ipa)和对苯二甲酸二甲酯(dmtp)等,所述二元醛为乙二醛、丙二醛等。当所述天然高分子化合物为海藻酸钠时,所述凝固剂为二价或三价金属离子化合物;当所述天然高分子化合物为壳聚糖、纤维素或蛋白质时,所述凝固剂为二元羧酸、尿素或二元醛。
17.根据上文技术方案,优选的情况下,所述乙醇或甲醇和水的体积比为1:0.5
‑
1:1.5,优选为1:1。
18.根据上文技术方案,优选的情况下,湿法纺丝过程中喷丝孔的孔径为0.5
‑
0.9mm,优选为0.7mm;挤出速率5
‑
15mm/min,优选为9mm/min。
19.根据上文技术方案,优选的情况下,将共混溶液纺制成纤维后,经水洗、牵伸和干燥得到成品纤维。
20.根据上文技术方案,优选的情况下,将混合溶液通过流延法成膜,干燥后进入凝固剂溶液中凝固成型,制成膜材料。
21.根据上文技术方案,优选的情况下,将共混溶液制成纤维后,经水洗、冷冻干燥后,得到纤维气凝胶。
22.根据上文技术方案,优选的情况下,冷冻干燥的温度-40~-55℃,压力为40~60pa,时间为24~48小时。
23.根据上文技术方案,优选的情况下,所述添加的方式为滴加,优选为逐滴滴加。
24.此外,本发明还涉及保护利用上述方法制备的无机纳米粒子动态交联双网络结构增强天然聚合物材料,包括纤维材料、膜材料、纤维气凝胶材料。
25.本发明提供了一种全新一步法无机纳米粒子动态交联双网络结构增强海藻酸钠纤维制备工艺,先将天然高分子化合物(如海藻酸钠(sa))溶解,待完全形成溶液后加入含双键有机单体(如丙烯酸(aa)、无机纳米粒子(vsnp)、引发剂(如aps),在氮气保护的条件下升高温度引发自由基聚合,使天然高分子化合物(如sa)与聚合反应过程中的含双键有机单体(如丙烯酸(aa))进行互穿,同时与vsnp发生动态交联反应。聚合反应进行到一定程度后加入到凝固剂(如cacl2)中,凝固剂(如ca
2+
)交联后的天然高分子化合物(如sa)与伪交联的聚丙烯酸(paa)形成dn结构,从而形成无机纳米粒子动态交联dn结构的海藻酸钠纤维或膜。
26.本发明的有益效果是:
27.通过添加无机纳米粒子增强网络是一种改性天然多糖聚合物新兴方法,这一改性方法通过聚合物和纳米颗粒之间相互作用形成增强网络来达到改性天然聚合物的目的。将无机纳米粒子添加到聚合物中通过弱作用力(如范德华力和氢键)或强作用力(如共价键、离子键和配位键)使两者结合,因加工过程中存在生成和解离等过程,从而可以动态交联,发生动态变化,可以显著增强聚合物的热稳定性、机械性能和尺寸稳定性。无机纳米粒子动态交联双网络结构增强天然聚合物纤维通常是以双网络结构为核心,将无机纳米粒子固定在已有的天然聚合物内制备而成,无机纳米粒子和双网络结构被定形在纤维内发生动态交联。对于无机纳米粒子和双网络结构动态交联类天然聚合物材料来说,无机纳米粒子和双网络结构促使天然多糖聚合物材料的动态交联和网络均化,增加了天然多糖聚合物材料的力学性能,拓宽了其应用领域。
28.本发明基于双网络结构设计,制备了具有无机纳米粒子vsnp动态交联的强韧型纤维材料,既包含天然高分子化合物(如sa)和含双键有机单体聚合得到的聚合物(如paa)分子链的互穿互锁,又包含vsnp与天然高分子化合物(如sa)、含双键有机单体聚合得到的聚合物(如paa)间的动态氢键和天然高分子化合物凝固交联形成的海藻酸钙(ca)中ca
2+
离子交联等非共价键的动态交联。硅基纳米粒子在有机相间狭长大孔缝隙中的动态氢键和双网络的互穿互锁骨架结构均可均化网络结构,并实现强迫互容。动态交联结构设计可避免化学交联型凝胶液弱凝胶效应对湿法纺丝和韧性的不利影响,形成完全的连续的非颗粒型骨架结构。通过键合引入硅基纳米粒子并动态交联有机双网络,提高杂化牢度,解决骨架连续性,增强骨架强度,降低密度,并省去硅基颗粒形成稳定连接长长的老化时间。协效动态氢键、离子交联、双网互锁等动态非共价牺牲键作用,在收缩及受力时实现能量耗散,增强增韧。选择以生物质原料为主、“一锅法”水体系加工和普通干燥条件,提高效率和环境友好性。无机纳米粒子被固载在双网络内,固载效果良好,且纤维的力学性能突出;制备工艺简单、常压、低转速和低温下即可很好成形,产率高;dn型交联结构力学性能较好,有效的改善现有产品的缺陷,可弥补传统湿法纺丝方法的断裂强度和断裂伸长率的不足,载体价格昂贵,稳定性差,流动性不好、宏观形态多为块状,应用范围单一等诸多不足,拓宽了无机纳米粒子和天然高分子纤维材的使用范围。
29.(1)本发明采用“一锅法”工艺制备,无机纳米粒子与aa和sa发生伪交联反应并被双网络结构互锁固载,生成具有双网络结构的纤维或膜材料。
30.(2)第一网络海藻酸钙(ca)与第二网络paa分子链互穿缠结形成双网络互锁结构,共同组成了纤维材料的骨架,其中无机纳米粒子vsnp引发aa发生自由基聚合,生成paa的柔性链结构。vsnp、paa的部分卷曲链结构和ca之间存在“动态牺牲键”提升天然高分子纤维材料的性能尤其是力学性能。
31.(3)传统湿法纺丝制备出ca纤维由于力学性能的缺陷其应用往往受到限制,本发明的无机纳米粒子动态交联双网络结构协效增强增韧海藻酸钠纤维大幅度地提高了海藻酸钠纤维的力学性能,将无机纳米粒子材料与双网络结构的优势合二为一,且可综合调控双网络组分的化学结构,调控微观和外观形态结构,实现其在天然高分子纤维材料的多方面应用,如纺织品、航天航空、体育用品、医用医疗等诸多领域。而且基于其化学结构和形态结构调控机理还可以选择不同双网络固载组分实现光、电等更多功能性。
32.本发明制得的天然高分子纤维材料性能优异且制备方法简单,所用原料均廉价易得,成本较低;天然高分子纤维材料保留了ca的第一网络结构,实现了纤维材料力学性能所需的骨架结构,为进一步功能化提供了结构设计的基础;本发明有效地提高天然聚合物纤维的力学性能,断裂强度达到3.68cn/dtex,断裂伸长率13.65%,相比于纯sa纤维分别提高了83.08%和65.45%,同样也可以增强天然聚合物膜的力学性能。无机纳米粒子动态交联双网络结构增强增韧效果明显,可应用于纺织品、航天航空、体育用品、医用医疗等诸多领域。
附图说明
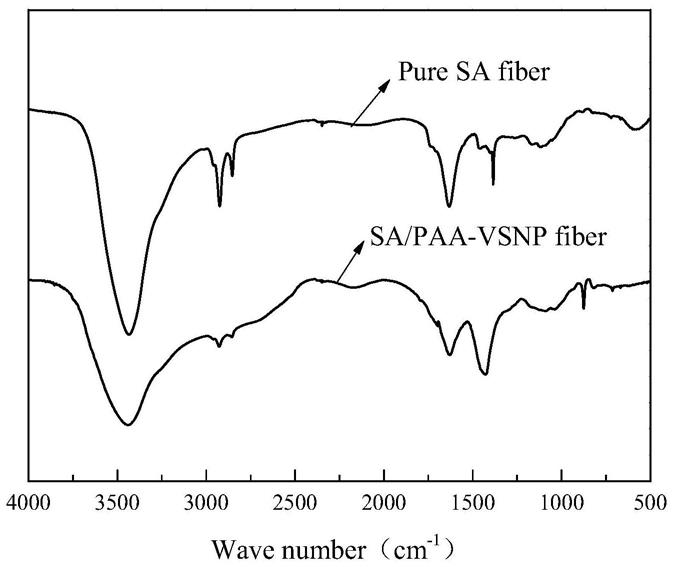
33.图1为实施例8制备的纤维材料的红外谱图。
34.图2为对比例1、实施例1、实施例2、实施例3、实施例4、实施例5制备的纤维的力学性能。
35.图3为对比例2、实施例2、实施例6、实施例7、实施例8、实施例9制备的纤维的力学性能。
36.图4为对比例2、实施例2、实施例8制备的纤维的sem图,其中a和a分别为对比例2纤维的表面和断面的sem图像,b和b分别为实施例2纤维的表面和断面的sem图像;c和c分别为实施例8纤维的表面和断面的sem图像。
37.图5为实施例8制备的纤维的eds图像。
38.图6为实施例1、实施例2、实施例3、实施例4、实施例5、实施例6、实施例7、实施例8、实施例9制备的纤维的tg和dtg(a和b:a、b、c、d和e分别代表paa的含量为10%,15%,20%,25%和30%;c和d:a、b、c、d和e分别代表vsnp的含量为1%,3%,5%,7%和9%)。
39.图7为实施例17制备的膜的接触角图像。
具体实施方式
40.下述非限定性实施例可以使本领域的普通技术人员更全面地理解本发明,但不以任何方式限制本发明。
41.下面结合附图和具体实例对本发明做进一步阐述。
42.下述实施例中海藻酸钠(sa)的分子量为5
×
106g/mol。
43.下述实施例中无机纳米粒子动态交联双网络结构增强海藻酸钠纤维的力学性能是通过ll
‑
06e型电子单纤强力测试仪进行测试的。
44.对比例1
45.(1)乙烯基三乙氧基硅烷(vtes)的水解
46.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
47.(2)sa
‑
vsnp共混溶液的制备
48.称2g sa加入到100ml烧杯中,配成2%sa溶液,在搅拌条件下,使用滴定器将0.3ml vsnp逐滴滴加到sa溶液中,在40℃水浴和氮气保护的条件下反应24h,得到sa
‑
vsnp共混溶液。
49.(3)sa
‑
vsnp纤维的制备
50.将sa
‑
vsnp纺丝液经变频步进器以9mm/min速率从孔径为0.7mm的喷丝孔挤出,进入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗、牵伸及室温干燥至恒重后得到sa
‑
vsnp纤维(paa:0%;vsnp:5%)。
51.实施例1
52.(1)乙烯基三乙氧基硅烷(vtes)的水解
53.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
54.(2)sa/paa
‑
vsnp共混溶液的制备
55.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.2ml aa单体和上述水解后的0.2ml vsnp逐滴滴加到sa溶液中,再加入0.004g aps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
56.(3)sa/paa
‑
vsnp纤维的制备
57.将sa/paa
‑
vsnp纺丝液经变频步进器以9mm/min速率从孔径为0.7mm的喷丝孔挤出,进入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗、牵伸及室温干燥至恒重后得到sa/paa
‑
vsnp纤维(paa:10%;vsnp:5%)。
58.实施例2
59.(1)乙烯基三乙氧基硅烷(vtes)的水解
60.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
61.(2)sa/paa
‑
vsnp共混溶液的制备
62.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.3ml vsnp逐滴滴加到sa溶液中,再加入0.006g aps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
63.(3)sa/paa
‑
vsnp纤维的制备
64.将sa/paa
‑
vsnp纺丝液经变频步进器以9mm/min速率从孔径为0.7mm的喷丝孔挤出,进入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗、牵伸及室温干燥至恒重后得到sa/paa
‑
vsnp纤维(paa:15%;vsnp:5%)。
aa单体逐滴滴加到sa溶液中,再加入0.006g aps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa共混溶液。
89.(2)sa/paa纤维的制备
90.将sa/paa纺丝液经变频步进器以9mm/min速率从孔径为0.7mm的喷丝孔挤出,进入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗、牵伸及室温干燥至恒重后得到sa/paa纤维(paa:15%;vsnp:0%)。
91.实施例6
92.(1)乙烯基三乙氧基硅烷(vtes)的水解
93.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
94.(2)sa/paa
‑
vsnp共混溶液的制备
95.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.06ml vsnp逐滴滴加到sa溶液中,再加入0.006gaps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
96.(3)sa/paa
‑
vsnp纤维的制备
97.将sa/paa
‑
vsnp纺丝液经变频步进器以9mm/min速率从孔径为0.7mm的喷丝孔挤出,进入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗、牵伸及室温干燥至恒重后得到sa/paa
‑
vsnp纤维(paa:15%;vsnp:1%)。
98.实施例7
99.(1)乙烯基三乙氧基硅烷(vtes)的水解
100.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
101.(2)sa/paa
‑
vsnp共混溶液的制备
102.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.18ml vsnp逐滴滴加到sa溶液中再加入0.006gaps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
103.(3)sa/paa
‑
vsnp纤维的制备
104.将sa/paa
‑
vsnp纺丝液经变频步进器以9mm/min速率从孔径为0.7mm的喷丝孔挤出,进入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗、牵伸及室温干燥至恒重后得到sa/paa
‑
vsnp纤维(paa:15%;vsnp:3%)。
105.实施例8
106.(1)乙烯基三乙氧基硅烷(vtes)的水解
107.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
108.(2)sa/paa
‑
vsnp共混溶液的制备
109.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.42ml vsnp逐滴滴加到sa溶液中再加入0.006gaps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
110.(3)sa/paa
‑
vsnp纤维的制备
111.将sa/paa
‑
vsnp纺丝液经变频步进器以9mm/min速率从孔径为0.7mm的喷丝孔挤出,进入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗、牵伸及室温干燥至恒重后得到sa/paa
‑
vsnp纤维(paa:15%;vsnp:7%)。
112.实施例9
113.(1)乙烯基三乙氧基硅烷(vtes)的水解
114.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
115.(2)sa/paa
‑
vsnp共混溶液的制备
116.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.54ml vsnp逐滴滴加到sa溶液中再加入0.006gaps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
117.(3)sa/paa
‑
vsnp纤维的制备
118.将sa/paa
‑
vsnp纺丝液经变频步进器以9mm/min速率从孔径为0.7mm的喷丝孔挤出,进入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗、牵伸及室温干燥至恒重后得到sa/paa
‑
vsnp纤维(paa:15%;vsnp:9%)。
119.实施例10
120.(1)乙烯基三乙氧基硅烷(vtes)的水解
121.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
122.(2)sa/paa
‑
vsnp共混溶液的制备
123.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.2ml aa单体和上述水解后的0.2ml vsnp逐滴滴加到sa溶液中,再加入0.004g aps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
124.(3)sa/paa
‑
vsnp膜的制备
125.使用30ml注射器抽取30ml sa/paa
‑
vsnp共混溶液挤入有框塑料培养皿中(直径=90mm,高度=14mm)中,然后将其放入40℃鼓风干燥箱中进行鼓风干燥5h,待其干燥后将其揭下放入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗并室温干燥至恒重后得到sa/paa
‑
vsnp膜(paa:10%;vsnp:5%)。
126.实施例11
127.(1)乙烯基三乙氧基硅烷(vtes)的水解
128.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
129.(2)sa/paa
‑
vsnp共混溶液的制备
130.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.3ml vsnp逐滴滴加到sa溶液中,再加入0.006g aps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
131.(3)sa/paa
‑
vsnp膜的制备
132.使用30ml注射器抽取30ml sa/paa
‑
vsnp共混溶液挤入有框塑料培养皿中(直径=90mm,高度=14mm)中,然后将其放入40℃鼓风干燥箱中进行鼓风干燥5h,待其干燥后将其
揭下放入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗并室温干燥至恒重后得到sa/paa
‑
vsnp膜(paa:15%;vsnp:5%)。
133.实施例12
134.(1)乙烯基三乙氧基硅烷(vtes)的水解
135.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
136.(2)sa/paa
‑
vsnp共混溶液的制备
137.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.4ml aa单体和上述水解后的0.4ml vsnp逐滴滴加到sa溶液中,再加入0.008g aps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
138.(3)sa/paa
‑
vsnp膜的制备
139.使用30ml注射器抽取30ml sa/paa
‑
vsnp共混溶液挤入有框塑料培养皿中(直径=90mm,高度=14mm)中,然后将其放入40℃鼓风干燥箱中进行鼓风干燥5h,待其干燥后将其揭下放入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗并室温干燥至恒重后得到sa/paa
‑
vsnp膜(paa:20%;vsnp:5%)。
140.实施例13
141.(1)乙烯基三乙氧基硅烷(vtes)的水解
142.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
143.(2)sa/paa
‑
vsnp共混溶液的制备
144.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.5ml aa单体和上述水解后的0.5ml vsnp逐滴滴加到sa溶液中,再加入0.01gaps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
145.(3)sa/paa
‑
vsnp膜的制备
146.使用30ml注射器抽取30ml sa/paa
‑
vsnp共混溶液挤入有框塑料培养皿中(直径=90mm,高度=14mm)中,然后将其放入40℃鼓风干燥箱中进行鼓风干燥5h,待其干燥后将其揭下放入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗并室温干燥至恒重后得到sa/paa
‑
vsnp膜(paa:25%;vsnp:5%)。
147.实施例14
148.(1)乙烯基三乙氧基硅烷(vtes)的水解
149.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
150.(2)sa/paa
‑
vsnp共混溶液的制备
151.称2gsa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.6ml aa单体和上述水解后的0.6ml vsnp逐滴滴加到sa溶液中,再加入0.012gaps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
152.(3)sa/paa
‑
vsnp膜的制备
153.使用30ml注射器抽取30ml sa/paa
‑
vsnp共混溶液挤入有框塑料培养皿中(直径=90mm,高度=14mm)中,然后将其放入40℃鼓风干燥箱中进行鼓风干燥5h,待其干燥后将其
揭下放入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗并室温干燥至恒重后得到sa/paa
‑
vsnp膜(paa:30%;vsnp:5%)。
154.实施例15
155.(1)乙烯基三乙氧基硅烷(vtes)的水解
156.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
157.(2)sa/paa
‑
vsnp共混溶液的制备
158.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.06ml vsnp逐滴滴加到sa溶液中,再加入0.006gaps。在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
159.(3)sa/paa
‑
vsnp膜的制备
160.使用30ml注射器抽取30ml sa/paa
‑
vsnp共混溶液挤入有框塑料培养皿中(直径=90mm,高度=14mm)中,然后将其放入40℃鼓风干燥箱中进行鼓风干燥5h,待其干燥后将其揭下放入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗并室温干燥至恒重后得到sa/paa
‑
vsnp膜(paa:15%;vsnp:1%)。
161.实施例16
162.(1)乙烯基三乙氧基硅烷(vtes)的水解
163.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
164.(2)sa/paa
‑
vsnp共混溶液的制备
165.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.18ml vsnp逐滴滴加到sa溶液中再加入0.006gaps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
166.(3)sa/paa
‑
vsnp膜的制备
167.使用30ml注射器抽取30ml sa/paa
‑
vsnp共混溶液挤入有框塑料培养皿中(直径=90mm,高度=14mm)中,然后将其放入40℃鼓风干燥箱中进行鼓风干燥5h,待其干燥后将其揭下放入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗并室温干燥至恒重后得到sa/paa
‑
vsnp膜(paa:15%;vsnp:3%)。
168.实施例17
169.(1)乙烯基三乙氧基硅烷(vtes)的水解
170.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
171.(2)sa/paa
‑
vsnp共混溶液的制备
172.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.42ml vsnp逐滴滴加到sa溶液中再加入0.006gaps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
173.(3)sa/paa
‑
vsnp膜的制备
174.使用30ml注射器抽取30ml sa/paa
‑
vsnp共混溶液挤入有框塑料培养皿中(直径=90mm,高度=14mm)中,然后将其放入40℃鼓风干燥箱中进行鼓风干燥5h,待其干燥后将其
揭下放入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗并室温干燥至恒重后得到sa/paa
‑
vsnp膜(paa:15%;vsnp:7%)。
175.实施例18
176.(1)乙烯基三乙氧基硅烷(vtes)的水解
177.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
178.(2)sa/paa
‑
vsnp共混溶液的制备
179.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.54ml vsnp逐滴滴加到sa溶液中再加入0.006gaps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
180.(3)sa/paa
‑
vsnp膜的制备
181.使用30ml注射器抽取30ml sa/paa
‑
vsnp共混溶液挤入有框塑料培养皿中(直径=90mm,高度=14mm)中,然后将其放入40℃鼓风干燥箱中进行鼓风干燥5h,待其干燥后将其揭下放入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后,经水洗并室温干燥至恒重后得到sa/paa
‑
vsnp膜(paa:15%;vsnp:9%)。
182.实施例19
183.(1)乙烯基三乙氧基硅烷(vtes)的水解
184.利用乙醇:去离子水=1:1的比例将1ml vtes稀释到5%,用醋酸调节上述溶液的ph=4~5,将所得溶液在室温下搅拌40min至澄清透明,得到水解产物vsnp。
185.(2)sa/paa
‑
vsnp共混溶液的制备
186.称2g sa加入到100ml烧杯中,配成2%sa溶液;在搅拌条件下,使用滴定器将0.3ml aa单体和上述水解后的0.42ml vsnp逐滴滴加到sa溶液中再加入0.006gaps,在40℃水浴和氮气保护的条件下反应24h,得到sa/paa
‑
vsnp共混溶液。
187.(3)sa/paa
‑
vsnp纤维气凝胶的制备
188.将sa/paa
‑
vsnp纺丝液经变频步进器以9mm/min速率从孔径为0.7mm的喷丝孔挤出,进入质量浓度为3%的cacl2溶液中凝固成型,凝固5min后进行水洗,在温度-50℃、压力为50pa的真空冷冻干燥机中放置48小时进行冷冻干燥,得到sa/paa
‑
vsnp纤维气凝胶(paa:15%;vsnp:7%)。
189.图1为实施例8制备的sa/paa
‑
vsnp纤维的红外谱图,从图1中可以看出,实施例8得到的sa/paa
‑
vsnp纤维与纯sa纤维相比,
‑
oh的伸缩振动峰变宽,并且发生红移,说明sa、paa和vsnp之间存在强烈的氢键作用。
190.图2为对比例1、实施例1、实施例2、实施例3、实施例4、实施例5制备的纤维的力学性能,从图2中可以看出,纤维的断裂强度和断裂伸长率随着paa含量的增加先增大后减小。当paa含量为15%时,sa/paa
‑
vsnp纤维的力学性能最佳。
191.图3为对比例2、实施例2、实施例6、实施例7、实施例8、实施例9制备的纤维的力学性能,从图3中可以看出,纤维的断裂强度随vsnp含量的增大而增大,断裂伸长率随着vsnp含量的增加先增大后减小。当vsnp含量为7%时,sa/paa
‑
vsnp纤维的力学性能最佳,其断裂强度达到3.68cn/dtex,断裂伸长率达到13.65%,相比于纯sa纤维分别提高了83.08%和65.45%。
192.图4为对比例2、实施例2、实施例8制备的纤维的sem图,其中a和a分别为对比例2纤维的表面和断面的sem图像,b和b分别为实施例2纤维的表面和断面的sem图像;c和c分别为实施例8纤维的表面和断面的sem图像;从图中可以看出,paa和vsnp引入体系后,纤维表面更加光滑并且纤维内部开始出现无机纳米粒子。
193.图5为实施例8制备的纤维的eds图像,从图5中可以看出ca和si均匀分布在纤维内部。
194.图6为实施例1、实施例2、实施例3、实施例4、实施例5、实施例6、实施例7、实施例8、实施例9制备的纤维的tg和dtg(a和b:a、b、c、d和e分别代表paa的含量为10%,15%,20%,25%和30%;c和d:a、b、c、d和e分别代表vsnp的含量为1%,3%,5%,7%和9%),从图6中可以看出随着paa和vsnp含量的增多,纤维的热性能不断提高。
195.图7为实施例17制备的膜的接触角图像,从图7中可以看出当paa含量为15%,vsnp含量为7%时,制备得到的膜材料的接触角为33.7
°
,具有强亲水性,这是因为随着vsnp的引入,大量的亲水性基团
‑
oh被引入到体系中。
196.对于任何熟悉本领域的技术人员而言,在不脱离本发明技术方案范围情况下,都可利用上述揭示的技术内容对本发明技术方案作出许多可能的变动和修饰,或修改为等同变化的等效实施例。因此,凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所做的任何简单修改、等同变化及修饰,均应仍属于本发明技术方案保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1