一种超细旦对位芳纶纤维制备工艺的制作方法
1.本发明属于化纤生产技术领域,涉及对位芳纶纤维制备工艺,具体是一种超细旦对位芳纶纤维制备工艺。
背景技术:
2.对位芳纶纤维具有高比强度、高比模量、耐高温和阻燃等优异性能,与碳纤维、高强高模聚乙烯并称为世界三大高性能纤维。对位芳纶具有良好的抗冲击、耐腐蚀和抗疲劳性能。
3.对位芳纶纤维一般由聚合工序,通过对苯二甲酰氯(tpc)和对苯二胺(ppd)经缩聚反应制得聚对苯二甲酰对苯二胺(ppta)粉料,再经过纺丝工序,将聚对苯二甲酰对苯二胺(ppta)粉料和浓硫酸按一定的配比加入到溶解机中,经过纺丝过程得到对位芳纶纤维。
4.专利文件(cn102560700a)公开了一种对位芳纶细旦纤维及其制备方法,采用高剪切双螺杆挤出机和连续薄膜脱泡装置,通过多道过滤工序,并通过脱泡纯净度以及喷丝温度、拉伸条件等,进行工艺整体优化,能够制得对位芳纶细旦纤维,但该方法由于存在原料配比工艺设置的缺陷,导致需要设置多道过滤工序,造成该方法制备对位芳纶细旦纤维工艺复杂。
5.专利文件(cn106906525a)公开了一种对位芳纶低旦纤维长丝的制备方法,该方法利用喷丝帽降低芳纶纺丝原液挤出漫流并丝的风险,用碱液洗涤初生丝束,快速中和硫酸,进而提高丝束强度,采用平铺式洗涤和干燥方式,降低洗涤干燥环节断丝的风险。该方法主要通过丝束洗涤工艺优化和丝束干燥工艺优化来提高对位芳纶低旦纤维丝的质量稳定性。但对并没有改影响丝束质量因素中纺丝原液的性质,因此该方法对提高对位芳纶低旦纤维的质量存在一定的局限性。
6.超细旦对位芳纶纤维和普通对位芳纶纤维纺丝流程的差异是低旦纤维丝束中纤维根数少,且单根纤维直径更小。因此,超细旦对位芳纶纤维对制备工艺要求严格,任何制备工艺控制点都会影响纺丝过程,任何工艺控制误差都会造成断丝,成品外观不良,最终影响客户的使用。
技术实现要素:
7.为了解决现有技术存在的问题,本发明提出一种超细旦对位芳纶纤维制备工艺,本发明的一种超细旦对位芳纶纤维制备工艺,包括以下步骤:s1将对苯二胺充分溶解在n-甲基吡咯烷酮/氯化钙溶液中,得到对苯二胺溶解物,得到的溶解物中,氯化钙的质量浓度为8.0%-8.6%。
8.s2在步骤s1得到的对苯二胺溶解物中第一次加入对苯二甲酰氯进行预聚反应,预聚反应降温后第二次加入对苯二甲酰氯,并在双螺杆反应器内进行聚合反应,得到聚合反应物;s3 将步骤s2得到的聚合反应物经过研磨、中和、洗涤、干燥后获得成品聚合物聚
对苯二甲酰对苯二胺;s4 将步骤s3得到的聚对苯二甲酰对苯二胺(ppta)粉料和浓硫酸按一定的配比加入到溶解机中,并控制反应初始温度,溶解完成后得到液晶态纺丝原液;s5 将步骤s4得到的液晶态纺丝原液经过脱泡,过滤后,进行喷丝;s6 将步骤s5喷丝后的对位芳纶纤维进行水洗、干燥后得到超细旦对位芳纶。
9.本发明中,所述加入的对苯二胺与对苯二甲酰氯的质量比为1:1,所述第一次加入对苯二甲酰氯与第二次加入对苯二甲酰氯的质量比为32%~37%:68%~63%;所述喷丝过程采用喷丝板,喷丝板孔数为20-100,纺织单丝为1d-4d的对位芳纶纤维。
10.本发明通过控制聚合双螺杆反应器的开度和对苯二胺与对苯二甲酰氯的配比及对苯二甲酰氯的加入方式,使得步骤s2得到的聚合物的iv值≥7,iv值为特性粘度值。
11.由于对苯二胺与对苯二甲酰氯的聚合反应是放热反应,因此如果一次性加入对苯二甲酰氯会导致聚合反应环境温度急剧变高,不利于苯二胺与对苯二甲酰氯聚合反应的完全进行,导致聚合反应不完全,产生大量副反应,另一方面,对苯二胺与对苯二甲酰氯聚合过程中放出大量的热,对反应设备要求较高,因此,本发明通过分步加入对苯二甲酰氯进行聚合反应,可确保对苯二胺与对苯二甲酰氯聚合反应的充分进行。
12.为了实现ppta和浓硫酸在溶解机中充分溶解,并形成光滑的液晶态纺丝原液,本发明步骤s2得到聚合反应物后,通过控制研磨机筛网粒径,使得到的ppta 粒径为200-450μm占比在90%以上,粒径分布呈正态分布,使得到的ppta为细小颗粒,如果主要粒径分布<200μm时,ppta为粉状,由于颗粒比表面积过大,硫酸不能完全浸润ppta,ppta不能完全溶解在硫酸中,最终形成的产物中存在大量的硫酸包覆团聚的ppta块状颗粒,块状颗粒会影响后续纺丝过程,如堵塞过滤器。当粒径>450μm时,由于ppta颗粒过大,会造成ppta颗粒内部无法与硫酸充分接触,无法完全溶解。
13.为了确保纺丝过程的顺利进行,本发明中,所述步骤s4得到的液晶态纺丝原液浓度为19.8%-20.5%,当液晶态纺丝原液浓度过低时,会导致丝束强度过低,断丝严重,当纺丝原液浓度过高,会导致无法正常进行纺丝。
14.由于ppta溶解和浓硫酸溶解是放热反应,因此,为了确保溶解过程的充分进行,本发明浓硫酸浓度为99.90%-100.00%,浓硫酸的初始温度控制在15℃-25℃。
15.所述步骤s4溶解过程,通过外部温度控制,使得反应初始温度为10℃-20℃。
16.所述步骤s4~s5的总时长为4h-5h。若总时长太长,会造成纺丝原液中,ppta和硫酸的沉降分层,纺丝过程生头困难。若时间太短,一方面会导致液晶态纺丝原液溶解不充分,可纺性差,另一方面纺丝原液中的so2/so3无法去除干净,纺丝原液中存留的so
2/
so3造成对芳纶纤维的品质造成影响。
17.所述步骤s6中,干燥前期温度为60-80℃,干燥后期温度为90-110℃,干燥时间均为0.5~1s。纤维经过水洗后的含有大量的水,为了将喷丝后的纤维含水量控制在3%-7%,并提高纤维的性能,本技术干燥前期温度为60-80℃,将纤维表面的水去除,干燥后期温度为90-110℃,将纤维中深层水烘出,进而起到烘干作用。若将两段温度均设置为60℃~80℃,由于纤维为皮芯结构,导致外层完成干燥后,但内层还保留大量水分;若将两段都设置为90-110℃,高温直接烘干纤维,对纤维表面造成不良缺陷,进而影响纤维性能。
18.所述步骤s5中喷丝过程所用的喷丝装置包括:入口法兰,所述入口法兰的一面设置液晶液喷嘴,入口法兰的另一侧为法兰面;与入口法兰的法兰面相连的底座,所述底座包括底座法兰面,设置在法兰面一端的空腔,所述空腔为空球状,所述底座空腔对应的外侧面设置外螺纹面;纺丝原液从喷嘴吐出,进入组件内部,后进行喷丝,作用连接卡套和原液管线。
19.所述底座空腔外侧设置分散板,所述分散板设置在空腔出口处,并与空腔过盈配合;原液通过泵输送,会产生一定的压力,分散板将压力分散开,稳定原液流量,将纺丝原液均匀分布到卡套内部。
20.与底座连接的喷丝板,所述喷丝板包括:底板,设置在底板上的圆柱体,由圆柱体另一端面形成的喷丝面,所述喷丝面的滤网孔为同心圆布局,并且喷丝面为由内向外的凹面,所述底板是由圆柱体端面外延形成的圆板;设置在底座外侧的卡套,所述卡套与喷丝板圆板直接设置密封垫圈,所述卡套内衬设置与底座配合的内螺纹面。
21.针对超细旦对位芳纶纤维,本技术设计相应的喷丝装置,选择喷丝板33、66孔喷丝板,纺织1-4d的超细旦对位芳纶纤维。
22.由于采用干喷湿纺工艺,依靠凝固浴托盘中的水流,制备初生纤维,通常情况下,最中间的液晶态原液的下落速度大于四周的液晶态原液的下落速度,因此会导致喷丝板内的液晶体喷丝不均匀,为了解决这一问题,本技术将喷丝板设置成凹面,从而缩短了中间液晶态原液和四周液晶态原液喷丝的时间差,降低喷丝装置内的液晶态原液喷出速度受工艺过程的影响,能够均匀喷丝。
23.本发明还提供了一种采用上述超细旦对位芳纶纤维制备工艺得到的超细旦对位芳纶纤维,所述超细旦对位芳纶纤维的外观平整,纤维旦数为1-4d,线密度偏差率<3%,断裂强度≥18cn/dtex,断裂伸长率为2%~4%,初始模量为70-110gpa,毛丝数量≤8个/2kg。本技术所述的毛丝数量的单位也可用烟台泰和新材料股份有限公司2017年12月28日 09点50分在企业标准信息公共服务平台(https://www.qybz.org.cn/)备案的《对位芳纶长丝》(q/0601 thx004-2017)中对位芳纶长丝的外观项目要求的单位个/筒,即本技术超细旦对位芳纶纤维的毛丝数量≤8个/筒。
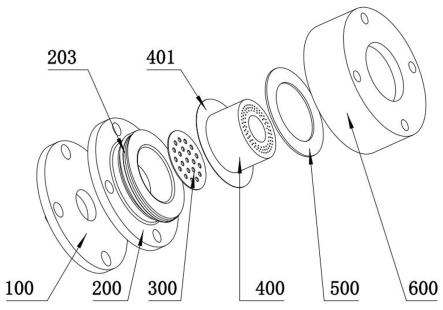
24.发明突出的技术效果为:由于对苯二胺与对苯二甲酰氯的聚合反应是放热反应,因此如果一次性加入对苯二甲酰氯会导致聚合反应环境温度急剧变高,不利于对苯二胺与对苯二甲酰氯聚合反应的完全进行,导致聚合反应不完全,产生大量副产物,另一方面,对苯二胺与对苯二甲酰氯聚合过程中放出大量的热,对反应设备要求较高,本发明通过分步加入对苯二甲酰氯进行聚合反应,可确保对苯二胺与对苯二甲酰氯聚合反应的充分进行。
25.本技术控制ppta粒径有利于得到最佳状态的纺丝原液,增加原液的流动性、光滑度,减少其中细小的硫酸包裹粉料的小疙瘩。
26.在ppta和浓硫酸混合过程中为放热反应,通过增加外部冷却系统,在进料螺杆设置夹套,ppta和浓硫酸混合时通过低温循环水,迅速将热量带走,控制反应初始温度为10℃-20℃,将ppta和浓硫酸混合过程产生的热量带走,可促进ppta和浓硫酸混合溶解过程液晶溶液的形成,降低因热量反应溶解剧烈过程造成ppta和硫酸溶解不充分,导致ppta和浓
硫酸溶解不充分形成ppta团聚硬块的问题。
27.本技术将纺丝原液从溶解制备到纺丝喷出整个过程的时间设置为4h-5h,使得原液停留时间设置为4h-5h,从而保证纺丝原液处于液晶状态,若纺丝液晶溶液停留时间过长,会造成纺丝原液中,ppta和硫酸的沉降分层,纺丝过程生头困难,纤维强度降低。若时间太短,一方面会导致液晶态纺丝原液溶解不充分,另一方面纺丝原液中的so2/so3无法去除干净,纺丝原液中存留的so2/so3无造成对芳纶纤维的s含量增加,进而影响纤维性能。控制溶解时间目的是得到流动性最佳的液晶态纺丝原液,有利于纺织细旦纤维。
28.由于干喷湿纺工艺过程,依靠凝固浴托盘中的水流,制备初生纤维,通常情况下,最中间的液晶态原液的下落速度大于四周的液晶态原液的下落速度,因此会导致喷丝板内的液晶体喷丝不均匀,为了解决这一问题,本技术将喷丝板设置成凹面,从而缩短了中间液晶态原液和四周液晶态原液喷丝的时间差,降低喷丝装置内的液晶态原液喷出速度受纺丝工艺的影响,能够均匀喷丝。超细旦纤维外观容易产生毛丝等瑕疵,影响客户使用,因此使用清水洗涤、减少张力辊,可有效避免因为设备导致的瑕疵。
附图说明
29.图1.喷丝装置的轴测图;图2.喷丝装置的轴测图的俯视图;图3 图2中a面剖视图;图中,入口法兰100,液晶液喷嘴101,法兰面102;底座200,底座法兰面201,空腔201,外螺纹面203,分散板300,喷丝板400,底板401,圆柱体402,喷丝面403,密封垫圈500,卡套600,内螺纹面601。
具体实施方式
30.下面实施例及对比例对本发明做进一步详细说明。
31.本发明的超细旦对位芳纶纤维制备工艺,包括以下步骤:s1将对苯二胺充分溶解在n-甲基吡咯烷酮/氯化钙溶液中,得到对苯二胺溶解物;s2在步骤s1得到的对苯二胺溶解物中第一次加入对苯二甲酰氯进行预聚反应,预聚反应降温后第二次加入对苯二甲酰氯,并在双螺杆反应器内进行聚合反应,得到聚合反应物;s3 将步骤s2得到的聚合反应物经过研磨、中和、洗涤、干燥后获得成品聚合物聚对苯二甲酰对苯二胺;s4 将步骤s3得到的聚对苯二甲酰对苯二胺和浓硫酸按一定的配比在溶解机中进行溶解,并控制反应初始温度,溶解完成后得到液晶态纺丝原液;s5 将步骤s4得到的液晶态纺丝原液经过脱泡,过滤后,进行喷丝;s6 将步骤s5喷丝后的对位芳纶纤维进行水洗、干燥后得到超细旦对位芳纶。
32.所述加入的苯二胺与对苯二甲酰氯的质量比为1:1,所述第一次加入对苯二甲酰氯与第二次加入对苯二甲酰氯的质量比为32%~37%:68%~63%;所述喷丝过程采用喷丝板,喷丝板孔数为20-100,纺织单丝为1d-4d的对位芳纶纤维。
33.所述步骤s2得到的聚合物的iv值≥7。
34.所述步骤s3中,通过控制研磨机筛网粒径,使得到的ppta80%的粒径为200-450μm。
35.所述步骤s4得到的液晶态纺丝原液浓度为19.8%-20.5%;所述步骤s4中,浓硫酸浓度为99.9%-100%,浓硫酸的初始温度控制在15℃-25℃;所述步骤s4溶解过程,通过外部温度控制,使得反应初始温度为10℃-20℃;所述步骤s5中,过滤器中滤网的大小3μm-10μm;所述步骤s4~s5的总时长为4h-5h;所述步骤s6中,干燥前期温度为60-80℃,干燥后期温度为90-110℃,干燥前期、干燥后期的干燥时间均为0.5~1s。
36.本技术的具体实施例如下:实施例1s1反应系统中持续通入对苯二胺溶解在n-甲基吡咯烷酮/氯化钙溶液中充分均匀混合,得到对苯二胺溶解物,通过控制对苯二胺与n-甲基吡咯烷酮/氯化钙溶液的通入量,使得对苯二胺溶解物中氯化钙的质量浓度为8.0%~8.3%;s2在步骤s1得到的对苯二胺溶解物中第一次加入32%对苯二甲酰氯进行预聚反应,预聚降温到5-6℃后第二次加入68%对苯二甲酰氯,两次加入的对苯二甲酰氯的总量与步骤s1得到的对苯二胺溶解物的质量比为1:1;并在双螺杆反应器内进行聚合反应,得到聚合反应物,本步骤中,得到的聚合物iv值为7.4;s3 将步骤s2得到的聚合反应物经过研磨、中和、洗涤、干燥后获得成品聚合物-聚对苯二甲酰对苯二胺ppta,本步骤控制研磨机筛网粒径,得到的ppta 90%的粒径为200-450μm;s4 将步骤s3得到的成品聚合物聚对苯二甲酰对苯二胺与浓度为99.9%的浓硫酸在溶解机中进行溶解,并控制反应初始温度为25℃,同时通过外部温度控制,使得反应初始温度为20℃,通过控制浓硫酸的加入量,得到溶解完成后浓度为19.8%液晶态纺丝原液;s5 将步骤s4得到的液晶态纺丝原液经过脱泡,过滤后,进行喷丝,过滤滤网的大小为3μm,喷丝过程采用喷丝板,喷丝板孔数为100,纺织单丝为1d;步骤s4-s5的总时长为4h;s6 将步骤s5喷丝后的对位芳纶纤维进行水洗、干燥后得到超细旦对位芳纶,干燥前期温度为80℃、干燥时间为0.5s,后期温度为110℃、干燥时间为0.5s。
37.对本实施例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率为2.4%,断裂强度为22.51cn/dtex,断裂伸长率为3.2%;初始模量为82gpa。
38.实施例2s1反应系统中持续通入苯二胺溶解在n-甲基吡咯烷酮/氯化钙溶液中充分均匀混合,得到混合物,得到对苯二胺溶解物,通过控制对苯二胺与n-甲基吡咯烷酮/氯化钙溶液的通入量,使得对苯二胺溶解物中氯化钙的质量浓度为8.2%~8.4%;s2在步骤s1得到的苯二胺溶解物中第一次加入 34%对苯二甲酰氯进行预聚反应,预聚降温到6-7℃后第二次加入66%对苯二甲酰氯,两次加入的对苯二甲酰氯的总量与步骤s1得到的对苯二胺溶解物的质量比为1:1,并在双螺杆反应器内进行聚合反应,得到聚合反应物;得到的聚合物iv值为7.2;
s3 将步骤s2得到的聚合反应物经过研磨、中和、洗涤、干燥后获得成品聚合物-聚对苯二甲酰对苯二胺ppta,本步骤控制研磨机筛网粒径,得到的ppta 90%的粒径为200-450μm;s4 将步骤s3得到的成品聚合物聚对苯二甲酰对苯二胺ppta与浓度为100%的浓硫酸在溶解机中进行溶解,并控制反应初始温度为15℃,同时通过外部温度控制,使得反应初始温度为10℃,通过控制浓硫酸的加入量,得到溶解完成后浓度为20.0%液晶态纺丝原液;s5 将步骤s4得到的液晶态纺丝原液经过脱泡,过滤后,进行喷丝,过滤滤网的大小为8μm,喷丝过程采用喷丝板,喷丝板孔数为50,纺织单丝为2d;步骤s4-s5的总时长为5h;s6 将步骤s5喷丝后的对位芳纶纤维进行水洗、干燥后得到超细旦对位芳纶,干燥前期温度为80℃、干燥时间为0.75s,后期温度为110℃、干燥时间为0.6s。
39.对本实施例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率为1.8%,断裂强度为23.84cn/dtex,断裂伸长率为3.4%;初始模量为95gpa。
40.实施例3s1反应系统中持续通入苯二胺溶解在n-甲基吡咯烷酮/氯化钙溶液中充分均匀混合,得到混合物,得到对苯二胺溶解物,通过控制对苯二胺与n-甲基吡咯烷酮/氯化钙溶液的通入量,使得对苯二胺溶解物中氯化钙的质量浓度为8.3%~8.5%;s2在步骤s1得到的苯二胺溶解物中第一次加入 35%对苯二甲酰氯进行预聚反应,预聚降温到 7-8℃后第二次加入65%对苯二甲酰氯,两次加入的对苯二甲酰氯的总量与步骤s1得到的对苯二胺溶解物的质量比为1:1,并在双螺杆反应器内进行聚合反应,得到的聚合物iv值为7.3;s3 将步骤s2得到的聚合反应物经过研磨、中和、洗涤、干燥后获得成品聚合物-聚对苯二甲酰对苯二胺ppta,本步骤控制研磨机筛网粒径,得到的聚对苯二甲酰对苯二胺ppta 90%的粒径为200-450μm;s4 将步骤s3得到的成品聚合物聚对苯二甲酰对苯二胺与浓度为99.9%的浓硫酸中在溶解机进行溶解,并控制反应初始温度为20℃,同时通过外部温度控制,使得反应初始温度为15℃,通过控制浓硫酸的加入量,得到溶解完成后浓度为20.2%液晶态纺丝原液;s5 将步骤s4得到的液晶态纺丝原液经过脱泡,过滤后,进行喷丝,过滤滤网的大小为10μm,喷丝过程采用喷丝板,喷丝板孔数为20,纺织单丝为4d;步骤s4-s5的总时长为4.5h;s6 将步骤s5喷丝后的对位芳纶纤维进行水洗、干燥后得到超细旦对位芳纶,干燥前期温度为70℃、干燥时间为0.8s,后期温度为100℃、干燥时间为0.8s。
41.对本实施例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率2.1%,断裂强度为23.57cn/dtex,断裂伸长率为3.5%;初始模量为83 gpa。
42.上述实施例3中,步骤s5中喷丝过程所用的喷丝装置包括:入口法兰100,所述入口法兰的一面设置液晶液喷嘴101,入口法兰的另一侧为法兰面102;与入口法兰的法兰面相连的底座200,所述底座200,包括底座法兰面201,设置在法兰面一端的空腔201,所述空腔为空球状,所述底座空腔对应的外侧面设置外螺纹面203;所述底座空腔外侧设置分散板300,所述分散板设置在空腔出口处,并与空腔过盈配合;与底座连接的喷丝板400,所述喷
丝板包括:底板401,设置在底板上的圆柱体402,由圆柱体另一端面形成的喷丝面403,所述喷丝面的滤网孔为同心圆布局,并且喷丝面为由内向外的凹面,所述底板是由圆柱体端面外延形成的圆板;设置在底座外侧的卡套600,所述卡套与喷丝板圆板直接设置密封垫圈500,所述卡套内衬设置与底座配合的内螺纹面601。
43.利用所述喷丝装置,设置不同的规格进行喷丝,其物理指标如表1:实施例4s1反应系统中持续通入苯二胺溶解在n-甲基吡咯烷酮/氯化钙溶液中充分均匀混合,得到混合物,得到对苯二胺溶解物,通过控制对苯二胺与n-甲基吡咯烷酮/氯化钙溶液的通入量,使得对苯二胺溶解物中氯化钙的质量浓度为8.4%~8.6%;s2在步骤s1得到的苯二胺溶解物中第一次加入37%对苯二甲酰氯进行预聚反应,预聚降温到8-10℃后第二次加入63%对苯二甲酰氯,两次加入的对苯二甲酰氯的总量与步骤s1得到的对苯二胺溶解物的质量比为1:1,并在双螺杆反应器内进行聚合反应,得到的聚合物iv值为7.3;s3 将步骤s2得到的聚合反应物经过研磨、中和、洗涤、干燥后获得成品聚合物-聚对苯二甲酰对苯二胺ppta ,本步骤控制研磨机筛网粒径,得到的聚对苯二甲酰对苯二胺ppta 90%的粒径为200-450μm;s4 将步骤s3得到的成品聚合物聚对苯二甲酰对苯二胺与浓度为100%的浓硫酸中在溶解机进行溶解,并控制反应初始温度为20℃,同时通过外部温度控制,使得反应初始温
度为15℃,通过控制浓硫酸的加入量,得到溶解完成后浓度为20.5%液晶态纺丝原液;s5 将步骤s4得到的液晶态纺丝原液经过脱泡,过滤后,进行喷丝,过滤滤网的大小为10μm,喷丝过程采用喷丝板,喷丝板孔数为20,纺织单丝为4d;步骤s4-s5的总时长为4.5h;s6 将步骤s5喷丝后的对位芳纶纤维进行水洗、干燥后得到超细旦对位芳纶,干燥前期温度为60℃、干燥时间为1s,后期温度为90℃、干燥时间为1s。
44.对本实施例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率2.2%,断裂强度为22.58cn/dtex,断裂伸长率为3.5%;初始模量为81 gpa。
45.对比例1本对比例与实施例3的不同仅在于步骤s2中,一次加入所有的对苯二甲酰氯后进行预聚,其他步骤及控制参数与实施例3相同,得到的聚合物iv值为6.8;对本对比例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率2.8%,断裂强度为18.23cn/dtex,断裂伸长率为2.8%;初始模量为81 gpa。
46.对比例2本对比例与实施例3的不同仅在于步骤s3中,控制研磨机筛网粒径,得到的聚对苯二甲酰对苯二胺ppta粒径90%分布为130μm-200μm;其他步骤及控制参数与实施例3相同。
47.在其他参数不变的情况下,粒径过低会导致硫酸不能完全浸润ppta,形成硫酸包裹粉料的小疙瘩。
48.对比例3本对比例与实施例3的不同仅在于步骤s3中,控制研磨机筛网粒径,得到的ppta粒径90% 为450μm-550μm;其他步骤及控制参数与实施例3相同。在其他参数不变的情况下,粒径>450μm时,由于ppta颗粒过大,会造成ppta颗粒内部无法与硫酸接触,无法完全溶解。
49.对比例4本对比例与实施例3的不同仅在于步骤s4中,没有对反应容器进行温度控制,其他步骤及控制参数与实施例3相同,由于ppta溶解在硫酸中的溶解反应是放热反应,由于没有外部控制温度,造成ppta和硫酸溶解不充分导致ppta溶解不充分形成ppta团聚硬块的问题。
50.对比例5本对比例与实施例3的不同仅在于步骤s4~s5的总时长为3.5h,其他步骤及控制参数与实施例3相同,对本对比例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率3.2%,断裂强度为17.24 cn/dtex,断裂伸长率为2.8%;初始模量为73 gpa。
51.反应时间太短,一方面会导致液晶态纺丝原液溶解不充分,另一方面纺丝原液中的so2无法去除干净,纺丝原液中存留的so2造成对芳纶纤维的品质造成影响,对比例6本对比例与实施例3的不同仅在于步骤s4~s5的总时长为5.5h,其他步骤及控制参数与实施例3相同,对本对比例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率4.3%。
52.纺丝液晶溶液停留时间过长,会造成纺丝原液中,ppta和硫酸的沉降分层,纤维线密度偏差大,纤维强度低。
53.对本对比例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率6.2%,断裂强度为10.76 cn/dtex,断裂伸长率为3.4%;初始模量为50gpa。
54.对比例7本对比例与实施例3的不同仅在于步骤s6纤维水洗后干燥温度设置为70℃,干燥时间为1.5s,其他步骤及控制参数与实施例3相同,对本对比例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率3.2%,断裂强度为20.33cn/dtex,断裂伸长率为2.5%;初始模量为73gpa。
55.对比例8本对比例与实施例3的不同仅在于步骤s6纤维水洗后干燥温度设置为100℃,干燥时间为1s,其他步骤及控制参数与实施例3相同,对本对比例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率3.3%,断裂强度为21.31cn/dtex,断裂伸长率为2.3%;初始模量为82gpa。
56.对比例9本对比例与实施例3的不同仅在于步骤s6中,干燥前期温度为50℃,干燥时间为1s,干燥后期温度为120℃,干燥时间为0.5s。
57.其他步骤及控制参数与实施例3相同,对本对比例得到的超细旦对位芳纶纤维进行材料性能检测,得到纤维的线密度偏差率4%,断裂强度为21.97cn/dtex,断裂伸长率为2%;初始模量为73gpa。
58.表2是上述不同实施例和对比例得到的纤维材料性能指标。
59.最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1