调色剂的制作方法
1.本发明涉及在使用电子照相法等记录方法中使用的调色剂。
背景技术:
2.近年来,例如复印机或打印机等图像形成设备在使用目的和使用环境方面多样化,并且要求在高速化、高图像品质化、和高稳定化方面得到改善。另外,复印机或打印机已经同时小型化或者在节能化方面得到改善。因此,优选使用利用在这些方面有用的磁性调色剂的磁性单组分显影系统。
3.电子照相法包括通过充电单元使静电潜像承载构件(以下,称为感光构件)带电的充电步骤,使带电的感光构件曝光以形成静电潜像的曝光步骤,和用调色剂使静电潜像显影以形成调色剂图像的显影步骤。接下来,通过经由中间转印体或未经由中间转印体而将调色剂图像转印到记录材料上的转印步骤,和通过使承载调色剂图像的记录材料通过由加压构件和可旋转图像加热构件形成的辊隙部来将调色剂图像加热和加压定影的定影步骤而将调色剂图像作为图像输出。特别地,在磁性单组分显影系统中,通过使用设置有例如磁性辊等磁场发生单元的调色剂载体(以下,称为套筒)来贮存磁性调色剂并且将磁性调色剂输送至显影区域来进行显影。
4.近年来,为了应对高图像品质化和节能化,各步骤的优化变得重要。为了改善图像品质,通常重要的是,优化用调色剂使静电潜像显影以形成调色剂图像的显影步骤和将调色剂图像从感光构件转印到记录材料的转印步骤。另外,为了改善节能,重要的是,在低温下进行充分定影。
5.考虑到涉及转印步骤的问题,转印缺陷可以是当转印期间存在问题时发生图像缺陷的实例。在转印步骤中,将转印偏压施加至感光构件上的调色剂并且通过静电引力使调色剂转印在记录介质上。此时,调色剂会残留在感光构件上而没有被转印,或者在转印期间可能会干扰调色剂层,并且由此,在图像上可能会发生缺陷或不均匀。该现象称为转印缺陷。
6.至今,为了改善转印性,已经尝试通过在抑制调色剂的流动性降低的同时,在调色剂生产时外部添加氧化铁(iron oxide)颗粒来进行测量(日本专利申请特开no.2000-214625和日本专利申请特开no.2005-37744)。
7.然而,在日本专利申请特开no.2000-214625和日本专利申请特开no.2005-37744中,在转印性和定影性之间的兼容性仍存在研究的空间。
8.具体地,在日本专利申请特开no.2000-214625和日本专利申请特开no.2005-37744中描述的调色剂中,没有获得充分的脱模性能,在定影步骤中,在加压构件、或图像加热构件等中可能会发生构件污染,这会引起图像缺陷。
9.另外,在例如用擦子(eraser)擦除写成的字符等将摩擦压力施加至定影图像的状态下,定影图像的调色剂会从纸剥离。
技术实现要素:
10.针对以上问题作出本发明。
11.即,本发明的一个目的在于,提供一种调色剂,其可以改善定影步骤中的脱模性能并且抑制构件污染,并且从图像剥离的调色剂的量少并且具有优异的摩擦定影性。
12.作为进行深入研究的结果,本发明人发现以上问题可以通过以下根据本发明的调色剂来解决。
13.即,根据本发明的调色剂是包括以下的调色剂:包含粘结剂树脂、脱模剂、和着色剂的调色剂颗粒;和存在于调色剂颗粒的表面上的氧化铁颗粒,其中氧化铁颗粒具有包含具有由下式(1)表示的结构的化合物的表面,
14.r-sio
3/2
(1)
15.其中,r表示碳原子数为1个以上的烃基。
16.参照附图,从以下示例性实施方案的描述,本发明的进一步特征将变得明显。
附图说明
17.图1是示出电容器模型的图。
18.图2示出通过观察进行了疏水化处理的氧化铁颗粒而获得的近边x射线吸收精细结构(nexafs)中si的吸收光谱的实例。
具体实施方式
19.现在将根据附图详细描述本发明的优选实施方案。
20.以下,将详细描述本发明,但本发明不限于此。
21.根据本发明的调色剂是包括以下的调色剂:包含粘结剂树脂、脱模剂、和着色剂的调色剂颗粒;和存在于调色剂颗粒的表面上的氧化铁颗粒,其中氧化铁颗粒具有包含具有由下式(1)表示的结构的化合物的表面,
22.r-sio
3/2
ꢀꢀ
(1)
23.其中,r表示碳原子数为1个以上的烃基。
24.在转印步骤中,在感光构件和记录介质之间使调色剂带电为正或负极性,并且将反转极性偏压施加至记录介质的背面上的转印材料。因为带电的调色剂颗粒沉积成几层,因此认为在转印步骤中发生沿着调色剂颗粒的表面的沿面放电。
25.在发生强的沿面放电的情况下,由于带电的无序,调色剂倾向于成为反转组分,这会引起其中记录介质上的调色剂返回到感光构件上的“再转印”。例如,当在输出实心黑色图像时频繁发生再转印时,转印缺陷变得显著,并且形成不均匀的图像。
26.即,为了抑制转印缺陷,需要抑制沿着调色剂表面的沿面放电。
27.此处,考虑到放电,可以想到图1中所示的电容器模型,其中感光构件和记录介质的背面上的转印材料用于电极板。假设电极11之间的电介质12为调色剂并且其电容为c,则c由以下等式表示。
28.c=εs/d
29.其中,s表示一个电极板的面积,d表示电极板之间的距离,和ε表示电极板之间的电介质的介电常数。
30.当施加在电极11之间的电场较大并且图1的电介质12的电容c小时,发生放电。
31.根据以上等式,电容c与作为电介质的调色剂的介电常数ε成比例。因此,期望具有高介电常数ε的调色剂以具有降低放电频率的效果。基于该想法,作为从具有高介电常数的物质的观点进行深入研究的结果,本发明人发现,在氧化铁颗粒存在于调色剂的表面上的情况下,可以获得显著效果。认为其原因是具有高介电常数的氧化铁颗粒存在于调色剂的表面上使得调色剂颗粒的表面上难以发生沿面放电。
32.另一方面,本发明人发现,在氧化铁颗粒存在于调色剂的表面上的情况下,在定影步骤中,加压构件或图像加热构件等中发生构件污染,这会引起图像缺陷。推测这是由于以下原因。
33.在外部添加氧化铁颗粒的情况下,转印性得到改善,但是氧化铁颗粒的表面是亲水性的,使得在调色剂中与脱模剂的亲和性低,并且在定影步骤中,脱模剂不容易在外部添加的氧化铁颗粒周围渗出。
34.通常,在定影步骤中,具有高急剧熔融性的脱模剂在图像表面上渗出。此时,当与脱模剂的亲和性低的氧化铁颗粒存在于调色剂的表面上时,脱模剂受排斥,使得形成其中图像表面的一部分未被脱模剂覆盖的区域。因此,认为在定影步骤中没有充分发挥脱模效果,导致构件污染。
35.由构件污染引起的图像缺陷在高速机器中显著发生。推测其原因涉及施加至调色剂层的热量。随着图像形成的速度越高,来自定影装置的热越难以传递至调色剂,并且大量不充分熔融的调色剂倾向于增多。即,其中从调色剂内部渗出的脱模剂的量不充分的调色剂的比例增多,并且更容易发生定影缺陷。
36.另外,在使用包含在表面上存在氧化铁颗粒的调色剂颗粒的调色剂形成定影图像,并且用擦子摩擦获得的定影图像的情况下,定影图像的调色剂可以容易地从纸剥离。推测这是因为脱模剂在具有亲水性表面的氧化铁颗粒的周围不具有亲和性,擦子的滑动性劣化,并且定影图像的调色剂由于擦子和调色剂之间的摩擦而从纸剥离。特别地,在纸纤维之间的纸的凹部处,在定影步骤中施加至调色剂的压力不充分,仅将调色剂的表面以熔融状态定影,并且从调色剂内部供给的脱模剂不充分。因此,擦子的滑动性显著劣化,并且调色剂容易从纸剥离。
37.在本发明中,对存在于调色剂颗粒的表面上的氧化铁颗粒进行疏水化处理,以便包含具有由式(1)表示的结构的化合物。即,存在于调色剂颗粒的表面上的氧化铁颗粒通过将具有碳原子数为1个以上的烃基的硅氧烷缩合物结合至氧化铁颗粒的表面而进行疏水化处理。因此,在使用根据本发明的调色剂的情况下,由于定影缺陷引起的构件污染得到抑制,并且摩擦定影性得到改善。
38.根据本发明人进行的研究,发现对氧化铁颗粒进行硅烷系表面处理,使得带电性得到改善并且显示调色剂的优异特性。在本发明中,改善带电性的类似效果可以通过用硅氧烷缩合物对氧化铁颗粒进行表面处理来获得。
39.另一方面,在使用例如除了si以外的钛酸酯系或铝酸酯系偶联剂缩合物等无机偶联剂缩合物的情况下,带电性比使用硅氧烷缩合物的情况更差。因此,在使用除了si以外的无机偶联剂缩合物进行表面处理的氧化铁颗粒存在于调色剂颗粒的表面上的情况下,调色剂的带电得到抑制,并且容易发生由于带电不均匀引起的沿面放电。结果,转印性不能与定
影性兼容。
40.另外,在用不具有r基的无机化合物对氧化铁颗粒进行表面处理的情况下,不显示疏水性,并且由此,不会获得改善脱模性能的效果,并且定影性无法得到改善。
41.另外,在式(1)的r基不是烃基的情况下,不会获得氧化铁颗粒的表面上的疏水性。因此,不会获得改善构件污染和摩擦定影性的效果。
42.此外,在使用硅油作为用于氧化铁颗粒的表面的疏水化处理的硅氧烷化合物的情况下,不能获得本发明的效果。推测这是因为硅油对氧化铁颗粒的固着率低,在定影步骤中硅油从氧化铁颗粒剥离,并且由此露出亲水性氧化铁表面。
43.式(1)中r基的碳原子数优选为1至20,更优选2至10,并且还更优选4至6。
44.式(1)的r基的碳原子数越少,氧化铁颗粒的表面和脱模剂之间的亲和性越低。因此,氧化铁颗粒附近吸引脱模剂的效果低,并且抑制由于定影缺陷引起的构件污染的效果也低。
45.另一方面,随着式(1)的r基的碳原子数增多,与脱模剂的亲和性提高,并且氧化铁颗粒附近吸引脱模剂的效果提高。然而,当碳原子数过大时,由于r基的位阻,使得氧化铁颗粒的表面不会均匀地疏水化。因此,在氧化铁颗粒的表面上产生未进行疏水化处理的区域,并且露出亲水性表面。结果,改善由于定影缺陷引起的构件污染和摩擦定影性的效果降低。
46.优选的是,氧化铁颗粒以充分粉碎的状态外部添加至调色剂的表面。在被覆在调色剂颗粒的表面上的氧化铁颗粒的二次聚集体的量大的情况下,在定影步骤中渗出的脱模剂受限于氧化铁颗粒的聚集颗粒中,并且不会充分获得氧化铁颗粒周围的脱模效果。氧化铁颗粒的粉碎状态可以通过用扫描电子显微镜测量调色剂表面上的氧化铁颗粒的聚集颗粒的直径来评价。氧化铁颗粒的聚集颗粒的直径优选为氧化铁颗粒的一次粒径的1.2倍以下。
47.氧化铁颗粒对调色剂颗粒的固着率优选为50至80%。当氧化铁颗粒的固着率为80%以下时,氧化铁颗粒包埋在调色剂的表面中,并且难以抑制脱模剂的渗出。另外,可以充分获得抑制转印步骤中调色剂表面上的沿面放电的效果,并且可以有效地抑制转印缺陷。另外,当氧化铁颗粒的固着率为50%以上时,氧化铁颗粒不太可能游离,并且由此,可以抑制转印缺陷的抑制效果的损失。
48.氧化铁颗粒对调色剂颗粒的固着率更优选为60至78%。
49.存在于调色剂颗粒的表面上的氧化铁颗粒相对于调色剂的总量的含量比优选为0.10至5.00质量%。当存在于调色剂颗粒的表面上的氧化铁颗粒相对于调色剂的总量的含量比为0.10质量%以上时,沿着调色剂层的表面的沿面放电得到显著抑制,并且转印性得到明显改善,并且由此,可以有效地抑制转印缺陷。另外,由于可以通过进行了疏水化处理的氧化铁颗粒来吸引在定影步骤中渗出的脱模剂,因此可以获得优异的脱模性能。另外,当存在于调色剂颗粒的表面上的氧化铁颗粒相对于调色剂的总量的含量比为5.00质量%以下时,氧化铁颗粒的量不过量,并且可以抑制通过游离的氧化铁颗粒对构件的磨损引起的白色条纹的出现。因此,可以抑制由于白色条纹的出现引起的实心黑色图像的图像浓度的降低。存在于调色剂颗粒的表面上的氧化铁颗粒相对于调色剂的总量的含量比更优选0.50至4.00质量%,并且还更优选1.00至2.50质量%。
50.在本发明中,粘结剂树脂的sp值和脱模剂的sp值之差优选为1.50以上,sp值通过
fedors法计算。另外,脱模剂的sp值和具有由式(1)表示的结构的化合物的sp值之差优选为1.20以下,sp值通过fedors法计算。
51.sp值是所谓的溶解度参数并且是用作表示多少物质溶解在某些物质中的溶解性或亲和性的指标的数值。当一种物质的sp值接近另一种物质的sp值时,该物质与另一种物质的溶解性或亲和性高,并且当一种物质的sp值远离另一种物质的sp值时,该物质与另一种物质的溶解性或亲和性低。在本发明中,sp值是基于常用的fedors法[poly.eng.sci.,14(2)147(1974)]计算的值。sp值的单位为(cal/cm3)
1/2
。
[0052]
为了使在定影步骤中加热和熔融的调色剂显示脱模效果,将脱模剂与粘结剂树脂分离并且渗出是必要的。另外,渗出的脱模剂需要均匀地铺展在调色剂表面上的氧化铁颗粒周围。
[0053]
当粘结剂树脂、脱模剂、和具有由式(1)表示的结构的化合物各自的sp值满足以上关系时,脱模剂可以在定影时从调色剂的内部渗出,以产生渗出的脱模剂铺展在调色剂表面上的氧化铁颗粒周围的状态。
[0054]
当粘结剂树脂的sp值和脱模剂的sp值之差为1.50以上时,脱模剂容易与粘结剂树脂分离并且渗出至调色剂的表面,由此容易显示脱模效果。
[0055]
另外,当脱模剂的sp值和具有由式(1)表示的结构的化合物的sp值之差为1.20以下时,脱模剂与在氧化铁颗粒的表面上的具有由式(1)表示的结构的化合物具有高亲和性。因此,可以充分解决渗出至调色剂表面的脱模剂难以渗出到未进行疏水化处理的氧化铁颗粒周围的问题。因此,可以获得高脱模效果,并且可以抑制由于构件污染引起的图像缺陷。
[0056]
粘结剂树脂的sp值和脱模剂的sp值之差更优选2.00至3.50,并且还更优选2.50至3.00。另外,脱模剂的sp值和具有由式(1)表示的结构的化合物的sp值之差更优选1.10以下,并且还更优选1.00以下。
[0057]
在本发明中,在通过使用差示扫描量热仪(dsc)、升温速度和降温速度二者均为100℃/分钟测量而获得的吸热曲线中,第二次升温过程中吸热峰的半值宽度优选为4.0至8.0℃。
[0058]
本发明中的吸热峰是指源自脱模剂的吸热峰。
[0059]
作为常规使用dsc的测量方法,例如,在根据jis k 7121(国际标准为astm d3418-82)的方法的情况下,升温速度通常作为10℃/分钟来测量。此处,重点在打印机的打印速度上,为了防止调色剂粘附至定影装置,需要例如,在几毫秒至几十毫秒的非常短的时间内调色剂熔融并且脱模剂渗出以便显示脱模效果。因此,作为集中深入研究脱模剂与粘结剂树脂分离并且渗出的能力的结果,本发明人发现将使用dsc的测量中升温速度和降温速度从通常的10改变为100℃/分钟是有效的。
[0060]
当测量的升温速度和降温速度各自为10℃/分钟时,考虑到脱模剂的移动,认为测量速度缓慢。即,在其中测量的升温速度和降温速度各自为10℃/分钟的测量条件下可以获得的参数中,假设几毫秒至几十毫秒作为调色剂可以从定影装置直接接收热的时间,则无法描述打印机在定影步骤中的脱模性能。
[0061]
接下来,将描述其中第一次升温和降温以及第二次升温全都以100℃/分钟进行的情况。
[0062]
在其中脱模剂和粘结剂树脂容易彼此相容的组合中,当第一次升温和冷却降温以
100℃/分钟进行时,没有时间将脱模剂与粘结剂树脂充分分离,并且在维持升温之后的熔融塑性状态的同时完成冷却。然后,当在维持塑性状态下以100℃/分钟再次升温时,在粘结剂树脂和脱模剂部分塑化的状态下升温,并且获得在粘结剂树脂和脱模剂彼此混合的状态下的吸热峰,而不是脱模剂特有的吸热峰。在该状态下,由于脱模剂和粘结剂树脂容易彼此相容,因此半值宽度更宽。
[0063]
在本发明中,第二次升温过程中吸热峰的半值宽度优选为4.0至8.0℃,并且意味着第二次升温过程中吸热峰的半值宽度在某种程度上是窄的。
[0064]
吸热峰的锐度反映了在升温过程中脱模剂的熔融和和随后与粘结剂树脂的分离进行的充分程度。即,在通过充分跟随快速升温速度而使脱模剂熔融并且进一步与粘结剂树脂分离的情况下,第二次升温过程中的吸热峰变得窄,并且半值宽度变成小的值。
[0065]
在本发明中,为了解决上述在定影步骤中构件污染,需要在几毫秒至几十毫秒的非常短的时间内使调色剂熔融和脱模剂渗出。因此,重要的是将第二次升温过程中吸热峰的半值宽度控制在预定范围内。
[0066]
在本发明中,当第二次升温过程中吸热峰的半值宽度为4.0℃以上时,粘结剂树脂和脱模剂的相在显影步骤中分离,并且调色剂表面上的脱模剂渗出,使得可以抑制显影性的劣化。另外,当第二次升温过程中吸热峰的半值宽度为8.0℃以下时,脱模剂的熔融或粘结剂树脂的分离可以充分跟随高速升温,并且在定影步骤中的构件污染可以得到抑制。
[0067]
在通过使用dsc、升温速度和降温速度二者均为100℃/分钟测量而获得的吸热曲线中,第二次升温过程中吸热峰的半值宽度更优选为4.0至6.0℃。
[0068]
在本发明中,存在于调色剂颗粒的表面上的氧化铁颗粒的润湿性优选为40至80vol%。存在于调色剂颗粒的表面上的氧化铁颗粒的润湿性如下确定。存在于调色剂颗粒的表面上的氧化铁颗粒以0.1g的量悬浮在50ml甲醇/水混合溶剂中,并且测量波长为780nm的光的透过率。然后,将当透过率为50%时甲醇浓度的值取作存在于调色剂颗粒的表面上的氧化铁颗粒的润湿性。存在于调色剂颗粒的表面上的氧化铁颗粒的润湿性更优选为55至60vol%。当润湿性在上述值的范围内时,氧化铁颗粒的带电性提高,并且与脱模剂的亲和性得到改善。因此,可以获得在氧化铁颗粒周围充分的脱模效果,并且由于构件污染引起的图像缺陷可以得到抑制。润湿性可以通过改变氧化铁颗粒的表面处理状态来控制。当润湿性为40vol%以上时,可以获得氧化铁颗粒表面的高程度的疏水化处理,并且可以获得在定影步骤中高脱模效果。另外,当润湿性为80vol%以下时,氧化铁颗粒包埋在调色剂颗粒的表面中可以得到抑制,并且例如由于调色剂的带电缺陷引起的转印缺陷或起雾等问题可以得到抑制。
[0069]
以下将描述水/甲醇润湿性测试方法。
[0070]
在本发明中,存在于氧化铁颗粒的表面上的具有由式(1)表示的结构的化合物优选为具有高缩合度的高度缩合的化合物。
[0071]
在存在高缩合状态的高分子量体的情况下,与存在低缩合状态的低分子量体的情况相比,疏水化处理剂倾向于更庞大(bulky)。因此,氧化铁颗粒表面上的烃链的密度提高,并且在定影步骤中与从调色剂的内部渗出的脱模剂的亲和性容易得到改善。结果,可以获得由于脱模缺陷引起的构件污染或摩擦定影性的改善的显著效果。
[0072]
以下将描述用于评价氧化铁颗粒的表面中所包含的具有由式(1)表示的结构的化
合物的缩合度的指标。
[0073]
在本发明中,[si-o-si]/[si-c]优选为1.4至1.7。此处,[si-o-si]/[si-c]如下定义。获得用甲苯从存在于调色剂颗粒的表面上的氧化铁颗粒提取的组分的红外吸收光谱(傅里叶变换红外(ft-ir)光谱)。在获得的ft-ir光谱中,在990至1,040cm-1
范围内的最大吸收峰强度定义为[si-o-si]。另外,在1,240至1,280cm-1
范围内的最大吸收峰强度定义为[si-c]。在该情况下,[si-o-si]与[si-c]的比为[si-o-si]/[si-c]。
[0074]
在进行了疏水化处理的氧化铁颗粒中,存在一定量的未结合至氧化铁颗粒的处理剂的缩合物。将氧化铁颗粒以100mg的量浸渍在50ml甲苯中,并且然后放置5小时,使得可以将处理剂的缩合物提取到甲苯中。疏水化处理剂的缩合状态可以通过测量通过除去氧化铁颗粒并且然后使甲苯挥发和干燥而获得的提取物的ft-ir光谱来确定。
[0075]
ft-ir光谱通过衰减全反射(atr)法来测量。在其中将ge用作atr晶体和红外光入射角为45
°
的条件下进行测量,以获得ft-ir光谱。在获得的ft-ir光谱中,将认为源自硅氧烷的si-o-si的、在990至1,040cm-1
范围内的最大吸收峰强度定义为[si-o-si]。另外,在获得的ft-ir光谱中,将认为源自硅氧烷的si-c的、在1,240至1,280cm-1
范围内的最大吸收峰强度定义为[si-c]。
[0076]
在获得通过将硅烷偶联处理剂水解而获得的单体单元中的ft-ir光谱的情况下,峰强度比[si-o-si]/[si-c]为1.3。
[0077]
另一方面,在获得通过将硅烷偶联处理剂的水解产物充分缩合而获得的多聚体中的ft-ir光谱的情况下,峰强度比[si-o-si]/[si-c]为1.7。
[0078]
当氧化铁颗粒的甲苯提取物中硅烷偶联剂的缩合率低时,由于提取物中单体单元的比例高,因此峰强度比[si-o-si]/[si-c]为接近1.3的值。另一方面,当氧化铁颗粒的甲苯提取物中硅烷偶联剂的缩合率高时,由于提取物中单体单元的比例低,因此峰强度比[si-o-si]/[si-c]为接近1.7的值。
[0079]
当[si-o-si]/[si-c]为1.4以上时,硅烷偶联剂的缩合率可以高,并且定影步骤中的脱模效果可以得到改善。
[0080]
以后将描述通过atr法的ft-ir光谱的测量。
[0081]
另外,在存在于调色剂颗粒的表面上的氧化铁颗粒中,当通过使用软x射线通过全电子产额(tey)法的测量来观察近边x射线吸收精细结构(以下,称为nexafs)时,获得的si的吸收光谱具有在1,844.4至1,844.8ev范围内的峰a和在1,846.1至1,846.6ev范围内的峰b,并且ia/(ia+ib)/msi优选为40至55g/mol,ia为峰a的面积,ib为峰b的面积,和msi为源自1g氧化铁颗粒中所包含的硅烷化合物的si的摩尔数。
[0082]
通过观察nexafs,可以获得结合至氧化铁颗粒的表面的硅烷化合物的si元素的结合状态的信息。nexafs通过其中在使用软x射线的光谱分析法中不选择从样品产生的电子的能量的全电子产额(tey)法来观察。
[0083]
软x射线穿透至距待测量的样品表面约50nm的深度,但是例如从样品表面溢出的光电子或俄歇电子等通过nexafs检测的电子限制在约5nm的深度。因此,可以非常显著地观察到氧化铁颗粒的表面上硅烷化合物的化学结合状态。
[0084]
图2示出通过测量进行了疏水化处理的氧化铁颗粒而获得的nexafs中si的吸收光谱的实例。si的吸收光谱在1,840至1,850ev的范围内具有两个峰a和b。峰a出现在低能量
侧,并且峰b出现在高能量侧。具体地,峰a的峰位出现在1,844.4至1,844.8ev的范围内,并且峰b的峰位出现在1,846.1至1,846.6ev的范围内。
[0085]
此处,已知当硅烷化合物中所包含的si原子和o原子之间的键由si-o-x表示时,当x为si时si原子和o原子之间的键对应于峰a,并且当x为fe时si原子和o原子之间的键对应于峰b。即,可以确定,结合至氧化铁颗粒的表面的硅烷化合物的量小,因为si的吸收光谱中峰a大,并且结合至氧化铁颗粒表面的硅烷化合物的量大,因为峰b大。
[0086]
由于化学结合至氧化铁颗粒的表面的硅烷化合物的比例小,硅烷偶联剂彼此高度缩合,并且氧化铁颗粒的表面上的硅烷化合物变得庞大。由于氧化铁颗粒的表面上的硅烷化合物变得庞大,与脱模剂的亲和性高,并且氧化铁颗粒周围的脱模效果得到进一步改善。
[0087]
即,测量上述nexafs中si的吸收光谱,使得可以获得氧化铁颗粒和硅烷偶联剂之间的结合比信息。因此,可以评价氧化铁颗粒的表面上的硅烷化合物的庞大(bulkiness)。
[0088]
当ia/(ia+ib)/msi为55g/mol以下时,硅烷化合物和氧化铁颗粒之间的结合强,并且亲水性氧化铁表面的露出得到抑制。当ia/(ia+ib)/msi为40g/mol以上时,硅烷化合物的缩合度可以高,并且可以获得高脱模效果。ia/(ia+ib)/msi更优选为43至48g/mol。
[0089]
用ia/(ia+ib)的值除以msi的值的原因是为了归一化。使用例如,扫描x射线荧光分光计zsx primusii(由rigaku corporation制造)来测量msi。
[0090]
根据本发明的调色剂包含存在于调色剂颗粒的表面上的氧化铁颗粒,对氧化铁颗粒进行疏水化处理,以便包含具有由式(1)表示的结构的化合物。此处,氧化铁颗粒可以通过外部添加至调色剂颗粒而包含在调色剂颗粒的表面上。
[0091]
氧化铁颗粒的实例包括例如磁铁矿、磁赤铁矿、或铁氧体等氧化铁;和例如铁、钴、或镍等金属或这些金属和例如铝、铜、镁、锡、锌、铍、钙、锰、硒、钛、钨、或钒等金属的合金,及其混合物。
[0092]
氧化铁颗粒的形状为八面体、六面体、球形、针状、或鳞片状等,并且可以使用任意形状,但是氧化铁颗粒的形状具有优选的例如至少四面体等多面体结构,并且更优选例如至少八面体等多面体结构。
[0093]
氧化铁颗粒的一次颗粒的数均粒径(d1)优选为0.50μm以下,并且更优选0.05至0.30μm。
[0094]
当氧化铁颗粒的一次颗粒的数均粒径(d1)为0.05至0.30μm时,在外部添加步骤中,氧化铁颗粒容易以一次颗粒的状态均匀附着至调色剂颗粒的表面,并且获得降低起雾的效果。氧化铁颗粒的一次颗粒的数均粒径(d1)更优选为0.10至0.30μm。
[0095]
另外,作为施加79.6ka/m时氧化铁颗粒的磁特性,当矫顽力(hc)为1.6至25.0ka/m时,显影性倾向于得到改善,这是优选的。矫顽力(hc)更优选为15.0至25.0ka/m。另外,磁化强度(σs)优选为30至90am2/kg,并且更优选40至80am2/kg。另外,剩余磁化(σr)优选为1.0至10.0am2/kg,并且更优选1.5至8.0am2/kg。
[0096]
氧化铁颗粒可以通过例如以下方法来生产。
[0097]
将相对于铁化合物等当量或等当量以上的例如氢氧化钠等碱添加至亚铁盐水溶液中,由此制备包含氢氧化亚铁的水溶液。在将制备的水溶液的ph维持在7以上的ph的同时,将空气吹入制备的水溶液中,并且在将水溶液加热至70℃以上的同时,进行氢氧化亚铁的氧化反应,使得首先生成氧化铁粉末的芯的晶种。
[0098]
接下来,将包含约1当量硫酸亚铁的水溶液添加至基于预先添加的碱的量包含晶种的浆状溶液中。在将溶液的ph维持在5至10并且吹入空气的同时,进行氢氧化亚铁的反应,由此使用晶种作为芯来生长氧化铁粉末。同时,可以通过选择任意的ph、反应温度、和搅拌条件来控制氧化铁颗粒的形状和磁特性。随着氧化反应进行,溶液的ph移动至酸性侧,并且优选的是,溶液的ph不小于5。氧化铁颗粒可以通过将通过常规方法如上所述获得的氧化铁颗粒过滤、洗涤、和干燥来获得。
[0099]
另外,在通过干法进行表面处理的情况下,对洗涤、过滤、和干燥的氧化铁颗粒进行偶联剂处理。当通过湿法进行表面处理时,在氧化反应之后将干燥的氧化铁颗粒再分散,或者在氧化反应之后,不干燥的情况下,将通过洗涤和过滤获得的氧化铁颗粒再分散在另一水系介质中,并且然后,进行偶联剂处理。在本发明中,可以适当地选择干法和湿法二者。
[0100]
可以在氧化铁颗粒的表面处理中使用的偶联剂的实例包括硅烷偶联剂。更优选使用具有由通式(2)表示的结构的硅烷偶联剂。
[0101]
r-sixnym(2)
[0102]
其中,x和y各自表示烷氧基,n和m各自独立地表示0至3的整数,n+m为3,和r表示烷基、苯基、乙烯基、环氧基、或(甲基)丙烯酸基。
[0103]
由通式(2)表示的硅烷偶联剂的实例包括乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、乙烯基三(β-甲氧基乙氧基)硅烷、β-(3,4-环氧环己基)乙基三甲氧基硅烷、γ-环氧丙氧基丙基三甲氧基硅烷、γ-环氧丙氧基丙基甲基二乙氧基硅烷、γ-氨基丙基三乙氧基硅烷、n-苯基-γ-氨基丙基三甲氧基硅烷、γ-甲基丙烯酰氧基丙基三甲氧基硅烷、乙烯基三乙酰氧基硅烷、甲基三甲氧基硅烷、二甲基二甲氧基硅烷、苯基三甲氧基硅烷、二苯基二甲氧基硅烷、甲基三乙氧基硅烷、二甲基二乙氧基硅烷、苯基三乙氧基硅烷、二苯基二乙氧基硅烷、正丙基三甲氧基硅烷、异丙基三甲氧基硅烷、正丁基三甲氧基硅烷、异丁基三甲氧基硅烷、三甲基甲氧基硅烷、正己基三甲氧基硅烷、正辛基三甲氧基硅烷、正辛基三乙氧基硅烷、正癸基三甲氧基硅烷、羟基丙基三甲氧基硅烷、正十六烷基三甲氧基硅烷、和正十八烷基三甲氧基硅烷。
[0104]
在本发明中,可以优选使用其中r为烷基的由通式(2)表示的硅烷偶联剂。其中,r优选为碳原子数为3至6的烷基,并且r特别优选为碳原子数为3或4的烷基。
[0105]
在使用硅烷偶联剂的情况下,可以单独将硅烷偶联剂用于处理,或者可以将多种硅烷偶联剂组合用于处理。当以组合使用多种硅烷偶联剂时,可以将各硅烷偶联剂用于处理,或者可以将全部硅烷偶联剂同时用于处理。
[0106]
用于处理的偶联剂的总量相对于100质量份氧化铁颗粒优选为0.9至3.0质量份,并且重要的是根据氧化铁颗粒的表面积、和偶联剂的反应性等调节处理剂的量。
[0107]
在本发明中,调色剂的粘结剂树脂的实例包括但不限于,乙烯基系树脂和聚酯系树脂,并且可以使用相关技术中已知的树脂。
[0108]
具体地,可以使用例如聚苯乙烯、苯乙烯-丙烯共聚物、苯乙烯-乙烯基甲苯共聚物、苯乙烯-丙烯酸甲酯共聚物、苯乙烯-丙烯酸乙酯共聚物、苯乙烯-丙烯酸丁酯共聚物、苯乙烯-丙烯酸辛酯共聚物、苯乙烯-甲基丙烯酸甲酯共聚物、苯乙烯-甲基丙烯酸乙酯共聚物、苯乙烯-甲基丙烯酸丁酯共聚物、苯乙烯-甲基丙烯酸辛酯共聚物、苯乙烯-丁二烯共聚物、苯乙烯-异戊二烯共聚物、苯乙烯-马来酸共聚物或苯乙烯-马来酸酯共聚物等苯乙烯系
共聚物,聚丙烯酸酯,聚甲基丙烯酸酯,和聚乙酸乙烯酯,并且这些树脂可以单独使用,或者可以使用其两种以上的组合。其中,在显影特性、和定影性方面,苯乙烯系共聚物和聚酯树脂是特别优选的。
[0109]
调色剂的玻璃化转变温度(tg)优选为40至70℃。当调色剂的玻璃化转变温度为40至70℃时,在维持优异的定影性的同时,可以改善贮存稳定性和耐久性。
[0110]
优选将电荷控制剂添加至根据本发明的调色剂中。作为负电荷控制剂,有机金属络合物和螯合物是有效的,并且其具体实例包括单偶氮金属络合物;乙酰丙酮金属络合物;和例如芳香族羟基羧酸或芳香族二羧酸等金属络合物。负电荷控制剂的市售产品的具体实例包括spilon black trh、t-77、和t-95(由hodogaya chemical co.,ltd.制造)以及bontron(注册商标)s-34、s-44、s-54、e-84、e-88、和e-89(由orient chemical industries co.,ltd.制造)。
[0111]
另外,正电荷控制剂的实例包括苯胺黑和脂肪酸金属盐改性的苯胺黑产物;例如三丁基苄基铵-1-羟基-4-萘磺酸盐或四丁基四氟硼酸铵等季铵盐,和例如其类似物的磷鎓盐等鎓盐,及其色淀颜料;三苯基甲烷染料及其色淀颜料(作为色淀剂,例如,磷钨酸、磷钼酸、磷钨钼酸、鞣酸、月桂酸、没食子酸、铁氰酸、或亚铁氰化物等);高级脂肪酸的金属盐;例如氧化二丁基锡、氧化二辛基锡、或氧化二环己基锡等氧化二有机锡;和例如硼酸二丁基锡、硼酸二辛基锡、或硼酸二环己基锡等硼酸有机锡。正电荷控制剂的市售产品的具体实例包括tp-302和tp-415(由hodogaya chemical co.,ltd.制造)、bontron(注册商标)n-01、n-04、n-07、和p-51(由orient chemical industries co.,ltd.制造)、和copy blue pr(由clariant ag制造)。
[0112]
这些电荷控制剂可以单独使用,或者以其两种以上的组合使用。从调色剂的带电量的观点,电荷控制剂的使用量相对于100质量份粘结剂树脂优选为0.1至10.0质量份,并且更优选0.1至5.0质量份。
[0113]
调色剂颗粒包含脱模剂。调色剂颗粒包含脱模剂,使得定影性得到改善。
[0114]
作为脱模剂,可以使用全部已知的脱模剂。其具体实例包括例如石蜡、微晶蜡、或凡士林等石油系蜡及其衍生物,褐煤蜡及其衍生物,通过费-托法获得的烃蜡及其衍生物,以聚乙烯或聚丙烯为代表的聚烯烃蜡及其衍生物,例如巴西棕榈蜡或小烛树蜡等天然蜡及其衍生物,和酯蜡。此处,衍生物包括氧化物、与乙烯基系单体的嵌段共聚物、或接枝改性产物。另外,作为酯蜡,可以使用单官能酯蜡、双官能酯蜡、或例如四官能或六官能酯蜡等多官能蜡。
[0115]
调色剂颗粒中脱模剂的含量比相对于100质量份粘结剂树脂优选为0.5至10质量份。当脱模剂的含量比在上述范围内时,定影性得到改善并且不损害调色剂的贮存稳定性。
[0116]
另外,可以通过其中在粘结剂树脂的生产中将树脂溶解在溶剂中、提高树脂溶液的温度、并且在进行搅拌的同时进行添加和混合的方法,或者其中在调色剂的生产中的熔融混炼期间进行添加的方法将脱模剂共混在粘结剂树脂中。
[0117]
脱模剂的用差示扫描量热仪(dsc)测量的最大吸热峰的峰值温度(以下,称为熔点)优选为60至140℃,并且更优选70至130℃。当最大吸热峰的峰值温度(熔点)为60至140℃时,调色剂容易在定影时塑化,并且定影性得到改善。另外,当长期贮存调色剂时,难以发生脱模剂的渗漏等,这是优选的。
[0118]
脱模剂的最大吸热峰的峰值温度可以使用差示扫描量热仪“q 1000”(由ta instruments制造)根据astm d3418-82来测量。在该情况下,将铟和锌各自的熔点用于设备的检测单元的温度校正,并且将铟的熔解热用于热量的校正。
[0119]
具体地,精确称量约10mg测量样品,将测量样品放入铝盘中,并且使用空铝盘作为参照,在30至200℃的测量温度和10℃/分钟的升温速度下进行测量。在测量中,一次升温至200℃,随后,以10℃/分钟降温至30℃,并且然后,以10℃/分钟再次升温。脱模剂的最大吸热峰的峰值温度从第二次升温过程中30至200℃的温度下的dsc曲线来确定。
[0120]
调色剂颗粒中所包含的着色剂的实例包括,但不特别限于,有机颜料、有机染料、和无机颜料,并且可以使用相关技术中已知的着色剂。
[0121]
青色系着色剂的实例包括铜酞菁化合物及其衍生物、蒽醌化合物、和碱性染料色淀化合物。具体地,青色系着色剂的实例包括以下:c.i.颜料蓝1、c.i.颜料蓝7、c.i.颜料蓝15、c.i.颜料蓝15:1、c.i.颜料蓝15:2、c.i.颜料蓝15:3、c.i.颜料蓝15:4、c.i.颜料蓝60、c.i.颜料蓝62、和c.i.颜料蓝66。
[0122]
品红色系着色剂的实例包括以下:缩合偶氮化合物、二酮吡咯并吡咯化合物、蒽醌化合物、喹吖啶酮化合物、碱性染料色淀化合物、萘酚化合物、苯并咪唑酮化合物、硫靛化合物、和苝化合物。具体地,品红色系着色剂的实例包括以下:c.i.颜料红2、c.i.颜料红3、c.i.颜料红5、c.i.颜料红6、c.i.颜料红7、c.i.颜料紫19、c.i.颜料红23、c.i.颜料红48:2、c.i.颜料红48:3、c.i.颜料红48:4、c.i.颜料红57:1、c.i.颜料红81:1、c.i.颜料红122、c.i.颜料红144、c.i.颜料红146、c.i.颜料红150、c.i.颜料红166、c.i.颜料红169、c.i.颜料红177、c.i.颜料红184、c.i.颜料红185、c.i.颜料红202、c.i.颜料红206、c.i.颜料红220、c.i.颜料红221、和c.i.颜料红254。
[0123]
黄色系着色剂的实例包括缩合偶氮化合物、异吲哚啉酮化合物、蒽醌化合物、偶氮金属络合物、次甲基化合物、和烯丙基酰胺化合物。具体地,黄色系着色剂的实例包括以下:c.i.颜料黄12、c.i.颜料黄13、c.i.颜料黄14、c.i.颜料黄15、c.i.颜料黄17、c.i.颜料黄62、c.i.颜料黄74、c.i.颜料黄83、c.i.颜料黄93、c.i.颜料黄94、c.i.颜料黄95、c.i.颜料黄97、c.i.颜料黄109、c.i.颜料黄110、c.i.颜料黄111、c.i.颜料黄120、c.i.颜料黄127、c.i.颜料黄128、c.i.颜料黄129、c.i.颜料黄147、c.i.颜料黄151、c.i.颜料黄154、c.i.颜料黄155、c.i.颜料黄168、c.i.颜料黄174、c.i.颜料黄175、c.i.颜料黄176、c.i.颜料黄180、c.i.颜料黄181、c.i.颜料黄185、c.i.颜料黄191、和c.i.颜料黄194。
[0124]
黑色系着色剂的实例包括炭黑和通过使用上述黄色系着色剂、品红色系着色剂、和青色系着色剂混合来颜色混合以得到黑色而提供的着色剂。
[0125]
这些着色剂可以单独使用,或作为混合物使用,或者以固溶体状态使用。从色相角、彩度、亮度、耐光性、ohp透明性、和调色剂颗粒中的分散性的观点,选择着色剂。
[0126]
在将磁性体用作着色剂的情况下,磁性体包含例如四氧化三铁或γ-氧化铁等磁性氧化铁作为主组分,并且可以包含例如磷、钴、镍、铜、镁、猛、铝、或硅等元素。磁性体的bet比表面积优选为2至30m2/g,并且更优选3至28m2/g,bet比表面积通过氮吸附法获得。另外,磁性体的摩氏硬度优选为5至7。磁性体的形状为多面体、八面体、六面体、球形、针状、或鳞片状等,并且在提高图像浓度方面,具有例如多面体、八面体、六面体、或球形等形状的低各向异性的磁性体是优选的。
[0127]
调色剂颗粒中着色剂的含量比相对于100质量份粘结剂树脂或构成粘结剂树脂的聚合性单体优选为1至20质量份。在将磁性粉末用作着色剂的情况下,调色剂颗粒中磁性粉末的含量比相对于100质量份粘结剂树脂或构成粘结剂树脂的聚合性单体优选为20至200质量份,并且更优选40至150质量份。
[0128]
根据需要,氧化铁颗粒可以添加至根据本发明的调色剂中,并且外部添加剂可以通过混合附着至调色剂的表面。
[0129]
外部添加剂的实例包括例如二氧化硅细颗粒、氧化铝细颗粒、二氧化钛细颗粒、氧化锌细颗粒、钛酸锶细颗粒、氧化铈细颗粒、和钛酸钙细颗粒等金属氧化物细颗粒(无机细颗粒)。另外,可以使用使用两种以上金属的复合氧化物细颗粒,并且可以以任意的组合使用选自这些细颗粒的两种以上。
[0130]
另外,树脂细颗粒或树脂细颗粒和无机细颗粒的有机-无机复合细颗粒可以用作外部添加剂。
[0131]
更优选的是,外部添加剂包括选自由二氧化硅细颗粒和有机-无机复合细颗粒组成的组中的至少一种。
[0132]
二氧化硅细颗粒的实例包括通过溶胶-凝胶法生产的溶胶-凝胶二氧化硅细颗粒、水性胶体二氧化硅细颗粒、醇性-二氧化硅细颗粒、通过气相法获得的气相法二氧化硅细颗粒、和熔融二氧化硅细颗粒。
[0133]
树脂细颗粒的实例包括例如乙烯基系树脂、聚酯树脂、和硅酮树脂等树脂颗粒。
[0134]
有机-无机复合细颗粒的实例包括由树脂细颗粒和无机细颗粒构成的有机-无机复合细颗粒。
[0135]
在有机-无机复合细颗粒的情况下,由无机细颗粒显示的优异的耐久性和带电性得到维持,并且在定影期间,由于具有低热容量的树脂材料的组分,因此难以阻碍调色剂颗粒的聚结并且难以引起定影阻碍。因此,容易实现耐久性和定影性二者。
[0136]
优选地,有机-无机复合细颗粒是具有由包埋在作为树脂组分的树脂细颗粒(优选地,乙烯基系树脂细颗粒)的表面中的无机细颗粒构成的凸部的复合细颗粒。更优选地,有机-无机复合细颗粒是具有其中无机细颗粒在乙烯基系树脂细颗粒的表面露出的结构的复合细颗粒。还更优选地,有机-无机复合细颗粒是具有其中乙烯基系树脂细颗粒的表面具有源自无机细颗粒的凸部的结构的复合细颗粒。
[0137]
构成有机-无机复合细颗粒的无机细颗粒的实例包括例如二氧化硅细颗粒、氧化铝细颗粒、二氧化钛细颗粒、氧化锌细颗粒、钛酸锶细颗粒、氧化铈细颗粒、和钛酸钙细颗粒等细颗粒。
[0138]
调色剂中外部添加剂的含量比相对于100质量份调色剂颗粒优选为0.1至20.0质量份。
[0139]
可以用疏水化处理剂对外部添加剂进行疏水化处理。
[0140]
疏水化处理剂的实例包括例如甲基三氯硅烷、二甲基二氯硅烷、三甲基氯硅烷、苯基三氯硅烷、二苯基二氯硅烷、叔丁基二甲基氯硅烷、和乙烯基三氯硅烷等氯硅烷类;
[0141]
例如四甲氧基硅烷、甲基三甲氧基硅烷、二甲基二甲氧基硅烷、苯基三甲氧基硅烷、二苯基二甲氧基硅烷、邻甲基苯基三甲氧基硅烷、对甲基苯基三甲氧基硅烷、正丁基三甲氧基硅烷、异丁基三甲氧基硅烷、己基三甲氧基硅烷、辛基三甲氧基硅烷、癸基三甲氧基
硅烷、十二烷基三甲氧基硅烷、四乙氧基硅烷、甲基三乙氧基硅烷、二甲基二乙氧基硅烷、苯基三乙氧基硅烷、二苯基二乙氧基硅烷、异丁基三乙氧基硅烷、癸基三乙氧基硅烷、乙烯基三乙氧基硅烷、γ-甲基丙烯酰氧基丙基三甲氧基硅烷、γ-环氧丙氧基丙基三甲氧基硅烷、γ-环氧丙氧基丙基甲基二甲氧基硅烷、γ-巯基丙基三甲氧基硅烷,γ-氯丙基三甲氧基硅烷,γ-氨基丙基三甲氧基硅烷,γ-氨基丙基三乙氧基硅烷,γ-(2-氨基乙基)氨基丙基三甲氧基硅烷、和γ-(2-氨基乙基)氨基丙基甲基二甲氧基硅烷等烷氧基硅烷类;
[0142]
例如六甲基二硅氮烷、六乙基二硅氮烷、六丙基二硅氮烷、六丁基二硅氮烷、六戊基二硅氮烷、六己基二硅氮烷、六环己基二硅氮烷、六苯基二硅氮烷、二乙烯基四甲基二硅氮烷、和二甲基四乙烯基二硅氮烷等硅氮烷类;
[0143]
例如二甲基硅油、甲基氢硅油、甲基苯基硅油、烷基改性硅油、氯烷基改性硅油、氯苯基改性硅油、脂肪酸改性硅油、聚醚改性硅油、烷氧基改性硅油、甲醇改性硅油、氨基改性硅油、氟改性硅油、和末端反应性硅油等硅油;
[0144]
例如六甲基环三硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷、六甲基二硅氧烷、和八甲基三硅氧烷等硅氧烷类;和
[0145]
例如,如十一烷酸、月桂酸、十三烷酸、十二烷酸、肉豆蔻酸、棕榈酸、十五烷酸、硬脂酸、十七烷酸、花生酸、褐煤酸、油酸、亚油酸、和花生四烯酸等长链脂肪酸,和这些脂肪酸与例如锌、铁、镁、铝、钙、钠、或钾等金属的盐等脂肪酸及其金属盐。
[0146]
其中,在容易疏水化处理方面,优选使用烷氧基硅烷类、硅氮烷类、和硅油。这些疏水化处理剂可以单独使用,或者可以使用其两种以上的组合。
[0147]
为了改善调色剂的流动性或带电性,调色剂可以包含多种外部添加剂。
[0148]
外部添加剂的一次颗粒的数均粒径优选为0.030至0.30μm。
[0149]
在根据本发明的调色剂中,可以以小量使用例如,如氟树脂粉末、硬脂酸锌粉末、或聚偏二氟乙烯粉末等润滑剂粉末等其它外部添加剂;例如氧化铈粉末、碳化硅粉末、或钛酸锶粉末等研磨剂;例如氧化钛粉末或氧化铝粉末等流动性赋予剂;防结块剂;和反极性有机细颗粒或无机细颗粒作为显影性改善剂,只要其不具有实质性的不利影响即可。此外,可以对外部添加剂的表面进行疏水化处理。
[0150]
调色剂颗粒的重均粒径(d4)优选为3.0至12.0μm,并且更优选4.0至10.0μm。当调色剂颗粒的重均粒径(d4)为3.0至12.0μm时,可以获得优异的流动性,并且可以对潜像充分进行显影。
[0151]
对调色剂的生产方法没有特别限制,并且可以采用已知的生产方法。调色剂的生产方法的实例包括粉碎法、聚合法、分散聚合法、缔合聚集法、溶解悬浮法、悬浮聚合法、和乳液聚集法。
[0152]
以下,将具体描述通过熔融混炼步骤和粉碎步骤生产调色剂的粉碎法,并且本发明不限于此。
[0153]
通过例如亨舍尔混合器或球磨机等混合机将例如,粘结剂树脂、着色剂、脱模剂、以及根据需要的电荷控制剂和外部添加剂彼此充分混合(混合步骤)。使用例如双螺杆挤出机、加热辊、混炼机或挤出机等加热混炼机将获得的混合物熔融混炼(熔融混炼步骤)。
[0154]
对获得的熔融混炼产物进行冷却和固体化,使用粉碎机进行粉碎(粉碎步骤),并且然后使用分级机进行分级(分级步骤),由此获得调色剂颗粒。此外,根据需要,通过例如
亨舍尔混合器等混合机将调色剂颗粒和外部添加剂彼此混合以获得调色剂。
[0155]
混合机的实例包括以下:fm混合器(由nippon coke&engineering co.,ltd.制造);super混合器(由kawata mfg.co.,ltd.制造);ribocone(由okawara mfg.co.,ltd.制造);nauta混合器、turbulizer、和cyclomix(由hosokawa micron corporation制造);spiral pin混合器(由pacific machinery&engineering co.,ltd.制造);和lodige混合器(由matsubo corporation制造)。
[0156]
加热混炼机的实例包括以下:krc混炼机(由kurimoto,ltd.制造);buss共混炼机(由buss co.,ltd.制造)、tem型挤出机(由toshiba machine machinery co.,ltd.制造);tex双螺杆混炼机(由the japan steel works,ltd.制造);pcm混炼机(由ikegai corp.制造);三辊磨、混合辊磨、和混炼机(由inoue mfg.,inc.制造);kneadex(由mitsui mining&smelting co.,ltd.制造);ms式加压混炼机和kneader-ruder(由moriyama manufacturing co.,ltd.制造);和班伯里混合器(由kobe steel,ltd.制造)。
[0157]
粉碎机的实例包括以下:counter喷射磨机、micron喷射机、和inomizer(由hosokawa micron corporation制造);ids型磨机和pjm喷射磨机(由nippon pneumatic mfg.co.,ltd.制造);交叉喷射磨机(由kurimoto,ltd.制造);ulmax(由nisso engineering co.,ltd.制造);sk jet o磨机(由seishin enterprise co.,ltd.制造);criptron(由kawasaki heavy industries,ltd.制造);涡轮增压机(由turbo kogyo co.,ltd.制造);和超级转子(由nisshin engineering inc.制造)。
[0158]
分级机的实例包括以下:classiel、micron分级机、和spedic分级机(由seishin enterprise co.,ltd.制造);涡轮分级机(由nisshin engineering inc.制造);微粉分离器、turboplex(atp)、和tsp分离器(由hosokawa micron corporation制造);弯头喷射机(由nittetsu mining co.,ltd.制造);分散分离器(由nippon pneumatic mfg.co.,ltd.制造);和ym microcut(由yasukawa shoji co.,ltd.制造).
[0159]
另外,用于筛分粗颗粒的筛分装置的实例包括以下:ultra sonic(由kouei-sangyou co.,ltd.制造);rezona筛和gyro筛(由tokuju corporation制造);vibrasonic系统(由dalton corporation制造);sonicreen(由sintokogio,ltd.制造);涡轮筛选器(由turbo kogyo co.,ltd.制造);超微粉碎机(由makino mfg.co.,ltd.制造);和圆形振动筛。
[0160]
从外部添加剂的分散性的观点,优选将外部添加步骤中的混合时间为调节为0.5至10.0分钟,并且更优选1.0至5.0分钟。
[0161]
接下来,将描述各物理性质的测量方法。
[0162]
《氧化铁颗粒的一次颗粒的数均粒径(d1)的测量方法》
[0163]
首先,用透射电子显微镜观察氧化铁颗粒。为了观察,可以使用例如,透射电子显微镜jem2800(由jeol,ltd.制造),以从拍摄的明场图像计算粒径。jem2800的图像拍摄条件如下。
[0164]
将待观察的氧化铁颗粒充分分散在环氧树脂中,并且然后在40℃的温度气氛下进行固化2天,由此获得固化产物。通过超薄切片机(由leica microsystems gmbh制造)将获得的固化产物用作薄片状样品。
[0165]
使用jem2800,在加速电压为200kv、放大倍率为100,000倍、和尺寸为1,024
×
1,024像素的条件下获得透射图像。
[0166]
使用图像分析软件“image-pro plus版本5.0”对获得的透射图像进行二值化,以测量100个氧化铁颗粒的长径,并且将算术平均值用作一次颗粒的数均粒径。
[0167]
《调色剂表面上的氧化铁颗粒的数均聚集粒径的测量方法》
[0168]
用扫描电子显微镜观察调色剂的表面,以测量氧化铁颗粒的数均聚集粒径。因此,可以观察到外部添加至调色剂表面的氧化铁颗粒的粉碎状态。
[0169]
为了观察,使用扫描电子显微镜s-4800(由hitachi high-tech corporation制造)从拍摄的反射电子图像计算粒径。s-4800的图像拍摄条件如下。
[0170]
(1)s-4800观察条件
[0171]
首先,s-4800的观察条件如下设定:加速电压:1.0kv,发射电流20μa,探测电流:正常,聚焦模式:uhr,和wd:3.0mm。
[0172]
检测器(u+bse模式)并且选择l.a.100,并且在放大至200,000倍的视场中观察反射电子图像。
[0173]
进行自动亮度调节,并且保存尺寸为1,280
×
960像素的图像。拍摄多个图像以获得其中可以分析至少100个氧化铁聚集颗粒的图像。
[0174]
(2)图像分析
[0175]
使用图像分析软件“image-pro plus版本5.0”对透射图像进行二值化,以测量100个氧化铁颗粒(聚集颗粒)的长径,并且将算术平均值用作氧化铁颗粒的数均聚集粒径。
[0176]
《存在于调色剂颗粒的表面上的氧化铁颗粒的定量方法》
[0177]
将包括氧化铁颗粒的外部添加剂组分从调色剂颗粒分离,并且将氧化铁颗粒进一步分离,并且从分离的外部添加剂组分回收,使得可以量化存在于调色剂颗粒的表面上的氧化铁颗粒。其具体方法的实例包括以下方法。
[0178]
(1)以5g的量将调色剂放入样品瓶中,并且向其中添加200ml甲醇。进一步,添加几滴“contaminon n”(包含非离子表面活性剂、阴离子表面活性剂、和有机助剂并且ph为7的10质量%精密测量仪器洗涤用中性洗涤剂的水溶液,由wako pure chemical industries,ltd.制造)。
[0179]
(2)用超声波清洁器分散样品5分钟,以分离外部添加剂组分。
[0180]
(3)通过抽吸过滤器(10μm膜过滤器)将调色剂颗粒和外部添加剂分离。
[0181]
(4)进行以上步骤(2)和(3)总计三次。
[0182]
通过以上操作,将外部添加剂组分与调色剂颗粒分离。将氧化铁颗粒分离并且用离心机通过回收溶液的离心分离来回收。接下来,除去溶剂并且通过真空干燥器将所得的颗粒充分干燥,以测量所得颗粒的质量,由此确定5g调色剂中氧化铁颗粒的含量。因此,可以确定调色剂中存在于调色剂颗粒的表面上的氧化铁颗粒的含量比。
[0183]
《氧化铁颗粒的固着率的测量方法》
[0184]
向50ml小瓶中称量20g“contaminon n”(包含非离子表面活性剂、阴离子表面活性剂、和有机助剂并且ph为7的10质量%精密测量仪器洗涤用中性洗涤剂的水溶液)并且与1g调色剂混合。
[0185]
将小瓶放入“km振动器”(型号:v.sx,由iwaki sangyo co.,ltd.制造)中,将速度设定为50,并且进行振动30秒。因此,将未固着的氧化铁颗粒从调色剂颗粒的表面转移至分散液。
[0186]
其后,通过离心机(商品名:h-9r,由kokusan co.,ltd.制造)以16.67s-1
将调色剂颗粒和转移至上清液中的氧化铁颗粒分散5分钟。将分离的调色剂真空干燥(40℃/24小时)以干燥和固体化,由此获得样品。
[0187]
通过压模机将调色剂造粒以用作样品。在以上处理前后的调色剂的样品中,待分析的氧化铁颗粒的特有fe元素通过以下波长色散x射线荧光(xrf)分析来定量化。然后,残留在调色剂颗粒的表面上而没有通过以上处理转移至上清液的氧化铁颗粒的量通过下式(a)来确定,并且将获得的值定义为固着率。作为固着率的值,采用从100个样品获得的值的算术平均值。
[0188]
(i)样品制备
[0189]
在样品的制备中,使用样品压模机maekawa试验机(由mfg co.,ltd.制造)。以0.5g的量将调色剂放入铝环(型号:3481e1)中,将载荷设定为5.0吨,并且按压调色剂1分钟,由此将调色剂造粒。
[0190]
(ii)使用的设备的实例
[0191]
x射线荧光分光仪3080(由rigaku corporation制造)
[0192]
(iii)测量条件
[0193]
测量直径:10
[0194]
测量电位和电压:50kv,50至70ma
[0195]
2θ角:25.12
°
[0196]
晶体板:lif
[0197]
测量时间:60秒
[0198]
(iv)氧化铁颗粒的固着率的计算方法
[0199]
氧化铁颗粒的固着率(%)=(源自处理后调色剂的氧化铁颗粒的元素的强度/源自处理前调色剂的氧化铁颗粒的元素的强度)
×
100
ꢀꢀ
(a)
[0200]
《sp值的计算方法》
[0201]
溶解度参数(sp值)使用由以下等式(b)表示的fedors等式来确定。
[0202]
δi=(ev/v)1/2=(δei/δvi)1/2
ꢀꢀ
(b)
[0203]
ev:蒸发能
[0204]
v:摩尔体积
[0205]
δei:组分i的原子或原子团的蒸发能
[0206]
δvi:组分i的原子或原子团的摩尔体积
[0207]
δei和δvi的值是指“基础涂料科学(basic coating science)”,54至57页,1986(maki shoten k.k.)的表3-9中描述的原子和原子团的蒸发能和摩尔体积(25℃)
[0208]
在等式(b)中,“δi”为组分i的原子或原子团的sp值,并且目标物质的sp值作为目标物质的原子或原子团的sp值δi之和而获得。
[0209]
在本发明中,具有由式(1)表示的结构的化合物的sp值是基于用于在氧化铁颗粒的表面上形成具有由式(1)表示的结构的化合物的疏水化处理剂的结构,使用式(b)计算的sp值。
[0210]
另外,sp值的单位为(cal/cm3)
1/2
并且可以通过1(cal/cm3)
1/2
=2.046
×
103(j/m3)
1/2
换算为单位(j/m3)
1/2
。
[0211]
《氧化铁颗粒的润湿性的测量方法》
[0212]
在使用水/甲醇混合溶剂的氧化铁颗粒的润湿性测试中,使用在以下条件和过程下使用粉末润湿性测试仪(商品名:wet-100p,由rhesca co.,ltd.制造)进行测量而获得的甲醇添加透过率曲线。
[0213]
首先,将50ml水放入烧瓶中,并且测量透过率。将此时的透过率设定为100%,并且将没有光透过的状态下的透过率设定为0%。然后,在将甲醇连续添加至水中以便增加甲醇浓度的同时,测量透过率。将当测量时透射光的强度为光穿过水时透射光强度的一半时的甲醇的浓度(质量%)定义为氧化铁颗粒的润湿性。
[0214]
具体地,透过率如下测量。
[0215]
将磁力搅拌器放入容纳有50ml水的烧杯中。然后,精确称量用开口为100μm的网筛选的0.1g氧化铁颗粒,并且放入烧瓶中。
[0216]
接下来,通过磁力搅拌器以300rpm(5转/秒)的搅拌速度开始搅拌,并且通过玻璃管以1.3ml/分钟的添加速度将甲醇连续添加至测量用样品溶液中。另外,此时,测量波长为780nm的光的透过率,以创建甲醇添加透过率曲线。此时,将甲醇用作滴定溶剂的原因是,用于处理氧化铁颗粒的表面的疏水化处理剂的洗脱的影响小并且可以更精确地评价氧化铁颗粒的表面性能。
[0217]
在测量中,例如,可以使用直径为5cm的玻璃烧杯作为烧杯,并且可以将涂覆有teflon(注册商标)的长度为25mm并且最大直径为8mm的纺锤形烧杯用作磁力搅拌器。
[0218]
《调色剂颗粒的重均粒径(d4)和数均粒径(d1)的测量》
[0219]
调色剂颗粒的重均粒径(d4)和数均粒径(d1)使用以下设备附带的专用软件、测量条件设定、和测量数据分析,用25,000个有效测量通道来测量,并且分析和计算测量的数据。
[0220]
·
设备:配备有100μm口管并且通过孔电阻法运行的精密粒径分布测量设备“coulter counter multisizer 3”(注册商标,由beckman coulter,inc.制造)
[0221]
·
专用软件:“beckman coulter multisizer 3version 3.51”(由beckman coulter,inc.制造)
[0222]
测量中使用的电解质水溶液通过将特级氯化钠溶解在离子交换水中以使得浓度为约1质量%来制备,并且可以使用例如,“isoton ii”(由beckman coulter,inc.制造)。
[0223]
在进行测量和分析之前,如下设定专用软件。
[0224]
在专用软件的“改变标准测量方法(som)屏幕”中,将控制模式的总计数设定为50,000个颗粒,将测量的次数设定为1,并且将通过使用“10.0μm标准颗粒”(由beckman coulter,inc.制造)获得的值设定为kd值。通过按下阈值/噪音水平的测量按钮来自动设定阈值和噪音水平。另外,将电流设定为1,600μa,将增益设定为2,并且将电解质溶液设定为isoton ii,并且勾选测量后冲洗口管。
[0225]
在专用软件的“脉冲至粒径转换设定屏幕”中,将元件间隔设定为对数粒径,将粒径元件设定为256个粒径元件,并且将粒径范围设定为2至60μm。
[0226]
具体测量方法如下。
[0227]
(1)将约200ml电解质水溶液放入专用于multisizer 3的250ml圆底玻璃烧杯中,将烧杯设置在样品台中,并且用搅拌棒以24转/秒在逆时针方向上进行搅拌。然后,通过专
用软件的“口管冲洗”功能除去口管中的污物和气泡。
[0228]
(2)将约30ml电解质水溶液放入100ml平底玻璃烧杯中。将约0.3ml通过用离子交换水稀释“contaminon n”(包含非离子表面活性剂、阴离子表面活性剂、和有机助剂并且ph为7的10质量%精密测量仪器洗涤用中性洗涤剂的水溶液,由wako pure chemical industries,ltd.制造)3质量倍而获得的稀释液作为分散剂添加至电解质水溶液中。
[0229]
(3)将预定量的离子交换水放入具有振荡频率为50khz、相偏移为180
°
的两个振荡器并且电力输出为120w的超声波分散器“ultrasonic dispersion system tetora 150”(由nikkaki bios co.,ltd.制造)的水槽中,并且将约2mlcontaminon n添加至水槽中。
[0230]
(4)将(2)中的烧杯设置在超声波分散器的烧杯固定孔中,并且启动超声波分散器。然后,调节烧杯的高度位置,使得烧杯中电解质水溶液的液面的共振状态最大化。
[0231]
(5)在用超声波照射(4)中的烧杯中的电解质水溶液的状态下,将约10mg调色剂(颗粒)逐渐添加至并且分散在电解质水溶液中。然后,超声波分散处理继续另外60秒。在超声波分散处理中,适当地调节水槽中的水温,使得温度为10℃至40℃。
[0232]
(6)使用移液管将(5)中的分散有调色剂颗粒的电解质水溶液滴入(1)中安装在样品台中的圆底烧杯中,以将测量浓度调节为约5%。然后,进行测量直到测量的颗粒数达到50,000为止。
[0233]
(7)用设备附带的专用软件分析测量数据以计算重均粒径(d4)。当用专用软件设定图表/体积%时,分析/体积统计值(算术平均)屏幕上的“平均直径”为重均粒径(d4)。另外,当用专用软件设定图表/数量%时,分析/数量统计值(算术平均)屏幕上的“平均直径”为数均粒径(d1)。
[0234]
《调色剂的吸热峰的半值宽度的测量方法》
[0235]
调色剂的吸热峰的半值宽度使用差示扫描量热仪“q 1000”(由ta instruments制造)如下测量。
[0236]
对于设备的检测单元的温度校准,使用铟和锌的熔点,并且对于热量的校准,使用铟的熔解热。
[0237]
作为测量样品,精确称量3.0mg调色剂并且将其放入铝盘中,并且将空的铝盘用作参照。
[0238]
在将温度保持在20℃下1分钟之后,在第一次升温过程中,在以100℃/分钟从30℃升温至200℃的同时,对测量样品进行测量。
[0239]
接下来,在将温度保持在200℃下1分钟之后,在以-100℃/分钟从200℃降温至30℃的同时,进行测量。
[0240]
最后,在将温度保持在30℃下1分钟之后,在以100℃/分钟从30℃升温至200℃的同时,在第二升温过程中进行测量。
[0241]
在第二升温过程中,在40至200℃的温度范围内获得比热变化,并且获得源自脱模剂的熔融的吸热峰。第二升温过程中脱模剂的熔点tm(℃)是比热变化曲线中最大吸热峰的峰值温度,并且吸热峰的半值宽度是最大吸热峰的峰值温度处的热量和基线之间的中点处的温度宽度。
[0242]
《测量ir和计算[si-o-si]/[si-c]值的方法》
[0243]
ft-ir光谱使用以下设备通过atr法来测量。
[0244]
·
配备有通用atr测量配件(universal atr sampling accessory)的傅里叶变换红外分光光度计(spectrum one,由perkinelmer inc.制造)
[0245]
具体测量步骤如下。
[0246]
将红外线(λ=5μm)的入射角设定为45
°
。将ge atr晶体(折射率:4.0)用作atr晶体。其它条件如下。
[0247]
范围
[0248]
开始:4000cm-1
[0249]
结束:600cm-1
[0250]
持续
[0251]
扫描次数:16
[0252]
分辨率:4.00cm-1
[0253]
[烃系蜡指数(ge)的计算方法]
[0254]
(1)将ge atr晶体安装在设备中
[0255]
(2)在atr晶体上,称量0.01g氧化铁颗粒的甲苯提取物。
[0256]
(3)用压力壁对样品加压,以测量。(测力计为90)
[0257]
(4)通过自动校正对获得的ft-ir光谱进行基线校正。
[0258]
(5)计算在990至1,040cm-1
范围内的最大吸收峰强度[si-o-si]与在1,240至1,280cm-1
范围内的最大吸收峰强度[si-c]的比[si-o-si]/[si-c]。
[0259]
根据本发明,可以提供可以改善定影步骤中的脱模性能并且抑制构件污染,并且从图像剥离的量少,并且摩擦定影性优异的调色剂。
[0260]
《实施例》
[0261]
以下,将参照生产例和实施例更具体地描述本发明,并且本发明不限于这些实施例。以下配方中的全部份数均为质量份。
[0262]
《氧化铁颗粒c1的生产例》
[0263]
在硫酸亚铁水溶液中,以如下所示的量将苛性钠溶液、p2o5、和sio2彼此混合,以制备包含氢氧化亚铁的水溶液。
[0264]
苛性钠溶液:相对于铁元素为1.00至1.10当量
[0265]
p2o5:相对于铁元素相当于以磷元素计为0.15质量%的量
[0266]
sio2:相对于铁元素相当于以硅元素计为0.50质量%的量
[0267]
将水溶液的ph设定为8.0,并且在吹入空气的同时在85℃下进行氧化反应,由此制备包含晶种的浆液。
[0268]
接下来,将硫酸亚铁水溶液添加至浆液中,使得其量相对于初始的碱量(苛性钠的钠组分)为0.90至1.20当量。其后,使浆液的ph维持在7.6,并且在吹入空气的同时使得进行氧化反应,由此获得包含氧化铁的浆液。
[0269]
进行过滤和洗涤,并且然后暂时取出含水浆液。此时,收集少量含水样品,并且测量水含量。
[0270]
接下来,将含水样品投入单独的水系介质中,而不干燥,并且在将浆料离心和搅拌的同时用离心磨进行再分散,由此将再分散液的ph调节至约9.0。然后,在进行搅拌的同时,以相对于100质量份氧化铁颗粒为1.4质量份(氧化铁颗粒的量作为从含水样品减去水含量
获得的值而计算)的量添加异丁基三甲氧基硅烷偶联剂,并且在45℃的液体温度下进行水解。其后,充分进行搅拌,并且进行表面处理。
[0271]
通过压滤机将进行疏水化处理而生产的氧化铁颗粒过滤,用大量水将过滤的颗粒洗涤,在120℃下将洗涤的颗粒干燥2小时,并且将获得的颗粒粉碎,由此获得数均粒径(d1)为0.26μm的氧化铁颗粒c1。
[0272]
《氧化铁颗粒c2至c8的生产例》
[0273]
在氧化铁颗粒c1的生产例中,将浆料的再分散液的初始ph设定为表1中所示的ph(a),添加表1中所示的疏水化处理剂,进行水解,将ph改变为表1中所示的ph(b),并且然后,进行表面处理。除了上述以外,通过与氧化铁颗粒c1的生产中相同的设备并且在相同的条件下获得氧化铁颗粒c2至c8。
[0274]
《氧化铁颗粒c9的生产例》
[0275]
在硫酸亚铁水溶液中,以如下所示的量将苛性钠溶液、p2o5、和sio2彼此混合,以制备包含氢氧化亚铁的水溶液。
[0276]
苛性钠溶液:相对于铁元素为1.00至1.10当量
[0277]
p2o5:相对于铁元素相当于以磷元素计为0.15质量%的量
[0278]
sio2:相对于铁元素相当于以硅元素计为0.50质量%的量
[0279]
将水溶液的ph设定为8.0,并且在吹入空气的同时在85℃下进行氧化反应,由此制备包含晶种的浆液。
[0280]
接下来,将硫酸亚铁水溶液添加至浆液中,使得其量相对于初始的碱量(苛性钠的钠组分)为0.90至1.20当量。其后,使浆液的ph维持在7.6,在吹入空气的同时使得进行氧化反应,在氧化反应最后将ph调节为6,并且进行洗涤和干燥。将获得的颗粒粉碎以获得数均粒径(d1)为0.23μm的氧化铁颗粒。
[0281]
作为疏水化处理剂,在进行搅拌的同时将30质量份异丁基三甲氧基硅烷滴加至70质量份离子交换水水中。其后,将水溶液保持在ph为5.5并且温度为55℃,并且使用分散叶片以0.46m/s的圆周速度分散120分钟,并且进行水解。其后,将水溶液的ph设定为7.0,并且将水溶液冷却至10℃以停止水解反应。由此,获得包含硅烷化合物的水溶液。
[0282]
在高速混合器(商品名:lfs-2型,由fukae powtec corporation制造)中,放入100质量份氧化铁颗粒,并且在以2,000rpm的旋转数进行搅拌的同时,在2分钟内滴加8.0质量份包含硅烷化合物的水溶液。其后,进行混合和搅拌5分钟。接下来,为了改善硅烷化合物的定影性,将混合物在40℃下干燥1小时,以减少水分的量,将混合物在110℃下干燥3小时,并且然后,使得进行硅烷化合物的缩合反应。其后,将缩合物粉碎,并且通过开口为100μm的网筛,由此获得氧化铁颗粒c9。
[0283]
《氧化铁颗粒c10至c16的生产例》
[0284]
在氧化铁颗粒c9的生产例中,并且如表1中所示改变所使用的疏水化处理剂的种类。除了上述以外,通过与氧化铁颗粒c9的生产中相同的设备并且在相同的条件下获得氧化铁颗粒c10至c16。
[0285]
《氧化铁颗粒c17的生产例》
[0286]
在硫酸亚铁水溶液中,以如下所示的量将苛性钠溶液、p2o5、和sio2(相对于铁元素相当于以硅元素计为0.50质量%的量)彼此混合,以制备包含氢氧化亚铁的水溶液。
[0287]
苛性钠溶液:相对于铁元素为1.00至1.10当量
[0288]
p2o5:相对于铁元素相当于以磷元素计为0.15质量%的量
[0289]
sio2:相对于铁元素相当于以硅元素计为0.50质量%的量
[0290]
将水溶液的ph设定为8.0,并且在吹入空气的同时在85℃下进行氧化反应,由此制备包含晶种的浆液。
[0291]
接下来,将硫酸亚铁水溶液添加至浆液中,使得其量相对于初始的碱量(苛性钠的钠组分)为0.90至1.20当量。其后,使浆液的ph维持在7.6,在吹入空气的同时使得进行氧化反应,并且在氧化反应最后将ph调节为6。其后,进行洗涤和干燥,并且将获得的颗粒粉碎,由此获得数均粒径(d1)为0.23μm的氧化铁颗粒。
[0292]
将氧化铁颗粒放入亨舍尔混合器(由nippon coke&engineering co.,ltd.制造)中,在以34.5m/s的转速分散未处理的氧化铁颗粒的状态下喷雾的同时,添加3.8质量份二甲基硅油,并且原样分散氧化铁颗粒10分钟。其后,获得通过开口为100μm的网筛的氧化铁颗粒作为氧化铁颗粒c17。
[0293]
《氧化铁颗粒c18的生产例》
[0294]
除了在氧化铁颗粒c17的生产例中,将疏水化处理剂的种类和使用量分别改变为改性硅油kf-415(由shin-etsu silicone co.,ltd.制造)和3.8质量份以外,通过与氧化铁颗粒c17的生产例中相同的设备并且在相同的条件下获得氧化铁颗粒c18。
[0295]
《氧化铁颗粒c19的生产例》
[0296]
在硫酸亚铁水溶液中,以如下所示的量将苛性钠溶液、p2o5、和sio2彼此混合,以制备包含氢氧化亚铁的水溶液。
[0297]
苛性钠溶液:相对于铁元素为1.00至1.10当量
[0298]
p2o5:相对于铁元素相当于以磷元素计为0.15质量%的量
[0299]
sio2:相对于铁元素相当于以硅元素计为0.50质量%的量
[0300]
将水溶液的ph设定为8.0,并且在吹入空气的同时在85℃下进行氧化反应,由此制备包含晶种的浆液。
[0301]
接下来,将硫酸亚铁水溶液添加至浆液中,使得其量相对于初始的碱量(苛性钠的钠组分)为0.90至1.20当量。其后,使浆液的ph维持在7.6,在吹入空气的同时使得进行氧化反应,在氧化反应最后将ph调节为6。其后,进行洗涤和干燥,并且将获得的颗粒粉碎,由此获得数均粒径(d1)为0.23μm的氧化铁颗粒。
[0302]
作为疏水化处理剂,在进行搅拌的同时将30质量份三异硬脂酰钛酸异丙酯滴加至70质量份离子交换水水中。其后,将水溶液保持在ph为5.5并且温度为55℃,并且使用分散叶片以0.46m/s的圆周速度分散120分钟,并且进行水解。其后,将水溶液的ph设定为7.0,并且将水溶液冷却至10℃以停止水解反应。由此,获得包含钛酸酯化合物的水溶液。
[0303]
在高速混合器(商品名:lfs-2型,由fukae powtec corporation制造)中,放入100质量份氧化铁颗粒,并且在以2,000rpm的旋转数进行搅拌的同时,在2分钟内滴加8.0质量份包含钛酸酯化合物的水溶液。其后,进行混合和搅拌5分钟。接下来,为了改善钛酸酯化合物的固着性,将混合物在40℃下干燥1小时,以减少水分的量,将混合物在110℃下干燥3小时,并且然后,使得进行钛酸酯化合物的缩合反应。其后,将缩合物粉碎,并且通过开口为100μm的网筛,由此获得氧化铁颗粒c19。
[0304]
《氧化铁颗粒c20的生产例》
[0305]
在硫酸亚铁水溶液中,以如下所示的量将苛性钠溶液、p2o5、和sio2彼此混合,以制备包含氢氧化亚铁的水溶液。
[0306]
苛性钠溶液:相对于铁元素为1.00至1.10当量
[0307]
p2o5:相对于铁元素相当于以磷元素计为0.15质量%的量
[0308]
sio2:相对于铁元素相当于以硅元素计为0.50质量%的量
[0309]
将水溶液的ph设定为8.0,并且在吹入空气的同时在85℃下进行氧化反应,由此制备包含晶种的浆液。
[0310]
接下来,将硫酸亚铁水溶液添加至浆液中,使得其量相对于初始的碱量(苛性钠的钠组分)为0.90至1.20当量。其后,使浆液的ph维持在7.6,在吹入空气的同时使得进行氧化反应,并且在氧化反应最后将ph调节至6。其后,进行洗涤和干燥,并且将获得的颗粒粉碎,由此获得数均粒径(d1)为0.23μm的氧化铁颗粒。
[0311]
接下来,将氧化铁颗粒添加至稀硫酸溶液中,以将ph调节至4。接下来,逐渐滴加硫酸铝水溶液,并且充分混合。在进一步继续进行搅拌的同时逐渐滴加氢氧化钠水溶液,以将悬浮液的ph调节至6,并且然后,使悬浮液老化。其后,进行洗涤和干燥,并且将获得的颗粒粉碎,由此获得数均粒径(d1)为0.23μm的氧化铁颗粒c20。
[0312]
《氧化铁颗粒c21的生产例》
[0313]
在氧化铁颗粒c9的生产例中,将疏水化处理剂的种类改变为四甲氧基硅烷。除了上述以外,通过与氧化铁颗粒c9的生产例中相同的设备并且在相同的条件下获得氧化铁颗粒c21。
[0314]
《氧化铁颗粒c22的生产例》
[0315]
在氧化铁颗粒c17的生产例中,将疏水化处理剂的种类和使用量分别改变为六甲基二硅氮烷和3.8质量份。除了上述以外,通过与氧化铁颗粒c17的生产例中相同的设备并且在相同的条件下获得氧化铁颗粒c22。
[0316]
《氧化铁颗粒c23的生产例》
[0317]
在硫酸亚铁水溶液中,以如下所示的量将苛性钠溶液、p2o5、和sio2彼此混合,以制备包含氢氧化亚铁的水溶液。
[0318]
苛性钠溶液:相对于铁元素为1.00至1.10当量
[0319]
p2o5:相对于铁元素相当于以磷元素计为0.15质量%的量
[0320]
sio2:相对于铁元素相当于以硅元素计为0.50质量%的量
[0321]
将水溶液的ph设定为8.0,并且在吹入空气的同时在85℃下进行氧化反应,由此制备包含晶种的浆液。
[0322]
接下来,将硫酸亚铁水溶液添加至浆液中,使得其量相对于初始的碱量(苛性钠的钠组分)为0.90至1.20当量。其后,使浆液的ph维持在7.6,在吹入空气的同时使得进行氧化反应,并且在氧化反应最后将ph调节至6。其后,进行洗涤和干燥,并且将获得的颗粒粉碎,由此获得数均粒径(d1)为0.23μm的氧化铁颗粒c23。氧化铁颗粒c23是未进行表面处理的氧化铁颗粒。
[0323]
[表1]
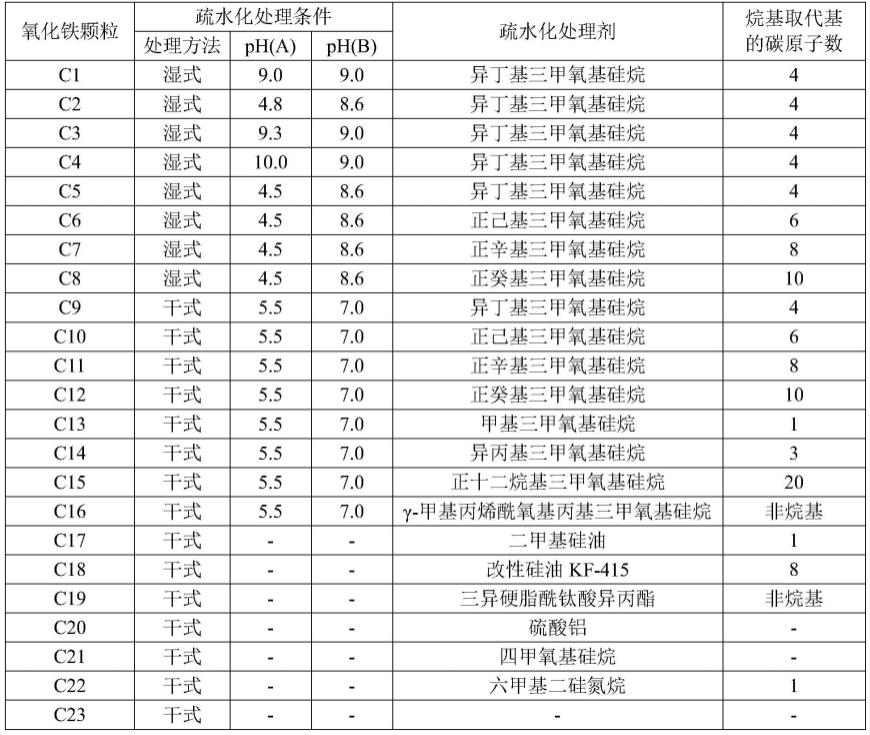
[0324][0325]
如上所述生产的氧化铁颗粒c1至c23的物理性质值示出在表2中,物理性质值通过以上方法来确定。
[0326]
[表2]
[0327][0328]
《磁性体(着色剂)的生产例》
[0329]
向92l的fe
2+
浓度为1.79mo1/l的硫酸亚铁水溶液中添加88l的3.74mo1/l氢氧化钠的水溶液,并且在以20l/分钟吹入空气的同时进行混合和搅拌,以便将温度和ph分别维持在89℃和9至12。在混合和搅拌30分钟之后,将浆料过滤、洗涤、并且干燥,以获得磁性体的颗粒。
[0330]
《调色剂颗粒a1的生产例》
[0331]
准备以下原料。
[0332]
·
非晶性聚酯树脂(pes)(通过双酚a的环氧乙烷和环氧丙烷加合物与对苯二甲酸的缩合反应获得的非晶性聚酯树脂,mw=9,500,tg=58℃)100质量份
[0333]
·
磁性体(着色剂)95质量份
[0334]
(数均粒径(d1):0.20μm,磁特性(σs:65.9am2/kg,σr:7.3am2/kg),未表面处理)
[0335]
·
脱模剂b1(山萮酸山萮酯,熔点:75℃)5.0质量份
[0336]
·
单偶氮染料的铁络合物(t-77,由hodogaya chemical co.,ltd.制造)2.0质量份
[0337]
通过亨舍尔混合器fm10c(由mitsui miike chemical engineering machinery co.,ltd.制造)将这些原料预混合。其后,通过将旋转数设定为250rpm并且设定温度调节为使得混炼产物的出口附近的直接温度为145℃的双螺杆混炼机(商品名:pcm-30,由ikegai corp.制造)将原料混炼。
[0338]
将获得的熔融混炼产物冷却,并且用切碎机将冷却的熔融混炼产物粗粉碎。因此,使用涡流t-250(由turbo kogyo co.,ltd.制造)以25kg/hr的进给速度,通过调节空气温度
使得排气温度为38℃将获得的粗粉碎产物细粉碎,并且使用利用附壁效应的多级分级机进行分级。结果,获得重均粒径(d4)为8.4μm的调色剂颗粒a1。
[0339]
《调色剂颗粒a2至a4的生产例》
[0340]
在调色剂颗粒a1的生产例中,如表3中所示改变使用的脱模剂的种类。除了上述以外,通过与调色剂颗粒a1的生产例中相同的设备并且在相同的条件下获得调色剂颗粒a2至a4。
[0341]
《调色剂颗粒a5的生产例》
[0342]
准备以下原料。
[0343]
·
苯乙烯/丙烯酸正丁酯共聚物1(stac)(苯乙烯丙烯酸系树脂(苯乙烯与丙烯酸正丁酯的质量比:78:22),mw=8,500,tg=58℃)100.0质量份
[0344]
·
磁性体(数均粒径(d1):0.20μm,磁特性(σs:65.9am2/kg,σr:7.3am2/kg),未表面处理)95.0质量份
[0345]
·
脱模剂b1(山萮酸山萮酯,熔点:75℃)5.0质量份
[0346]
·
单偶氮染料的铁络合物(商品名:t-77,由hodogaya chemical co.,ltd.制造)2.0质量份
[0347]
通过与调色剂颗粒a1的生产例中相同的设备并且在相同的条件下处理这些原料,由此获得调色剂颗粒a5。
[0348]
《调色剂颗粒a6至a8的生产》
[0349]
在调色剂颗粒a5的生产例中,如表3中所示改变使用的脱模剂。除了上述以外,通过与调色剂颗粒a5的生产例中相同的设备并且在相同的条件下获得调色剂颗粒a6至a8。
[0350]
《调色剂颗粒a9的生产例》
[0351]
根据以下步骤通过乳液聚集法来生产调色剂颗粒a9。
[0352]
将苯乙烯、丙烯酸丁酯、丙烯酸、和正月桂基硫醇混合并且分别以89.5份、9.2份、1.3份、和3.2份的量溶解。通过将1.5份neogen rk(由dks co.,ltd.制造)与150份离子交换水混合来制备水溶液,并且将混合物添加至上述制备的混合溶液中并且分散在其中。
[0353]
通过将0.3份过硫酸钾与10份离子交换水混合来制备过硫酸钾水溶液。在缓慢搅拌10分钟的同时添加过硫酸钾水溶液。
[0354]
在氮气置换之后,在70℃下进行乳液聚合6小时。在聚合完成之后,将反应溶液冷却至室温并且添加离子交换水,由此获得固成分浓度为12.5质量%并且基于体积的中值直径为0.2μm的粘结剂树脂颗粒分散液。
[0355]
将脱模剂(100份)(费-托蜡,熔点:77℃)和neogen rk(15份)混合在385份离子交换水中,并且使用湿式喷射磨机jn100(由jokoh co.,ltd.制造)将混合物分散约1小时,由此获得脱模剂分散液。脱模剂分散液的固成分浓度为20质量%。
[0356]
将磁性氧化铁颗粒(100份)和neogen sc(10.0份)混合在890份离子交换水水中,并且使用湿式喷射磨机jn100将混合物分散约1小时,由此获得磁性氧化铁分散液。
[0357]
将粘结剂树脂颗粒分散液(265份)、脱模剂分散液(10份)、和磁性氧化铁分散液(65份)放入容器中,并且使用均质器(商品名:ultra turrax t50,由ika works inc.制造)将混合物分散。
[0358]
在搅拌混合物的同时将容器中的温度调节至30℃,并且添加1mol/l盐酸以将ph调
节至5.0。放置3分钟之后,开始升温,并且升温至50℃以生产聚集颗粒。在该状态下,聚集颗粒的粒径通过“coulter counter multisizer 3”(注册商标,由beckman coulter,inc.制造)来测量。当聚集颗粒的重均粒径为6.2μm时,添加1mol/l氢氧化钠水溶液,并且将ph调节至8.0,以停止颗粒生长。
[0359]
其后,升温至95℃以将聚集颗粒熔融和球形化。当平均圆形度达到0.980时,开始降温,并且降温至30℃,由此获得调色剂颗粒分散液。
[0360]
将盐酸添加至获得的调色剂颗粒分散液中,以将ph调节至1.5以下,并且将混合物搅拌并且放置1小时,并且然后用压滤器进行固液分离,由此获得调色剂滤饼。
[0361]
用离子交换水将调色剂滤饼再浆化为分散液,并且用上述过滤器对分散液进行固液分离。重复再浆化和固液分离,直到滤液的电导率为5.0μs/cm以下为止,并且然后进行最终的固液分离,由此获得调色剂滤饼。
[0362]
用气流干燥器(气流喷射干燥器,由seishin enterprise co.,ltd.制造)将获得的调色剂滤饼干燥。在吹气温度为90℃的条件下进行干燥。干燥器出口温度为40℃,调节调色剂滤饼供给速度,使得出口温度不依赖调色剂滤饼的水含量而偏离40℃。
[0363]
此外,使用利用附壁效应的多级分级机切割微细的粗粉末,以获得调色剂颗粒a9。调色剂颗粒a9的重均粒径(d4)为8.4μm,调色剂颗粒a9的平均圆形度为0.980,并且调色剂颗粒a9的玻璃化转变温度(tg)为57℃。
[0364]
《调色剂颗粒a10至a12的生产例》
[0365]
在调色剂颗粒a5的生产例中,如表3中所示改变脱模剂的种类和使用量。除了上述以外,通过与调色剂颗粒a5的生产例中相同的设备并且在相同的条件下获得调色剂颗粒a10至a12。
[0366]
《调色剂颗粒a13的生产例》
[0367]
在调色剂颗粒a4的生产例中,如表3中所示改变使用的脱模剂的使用量。除了上述以外,通过与调色剂颗粒a4的生产例中相同的设备并且在相同的条件下获得调色剂颗粒a13。
[0368]
用于生产调色剂颗粒a1至a13的脱模剂的种类和通过上述方法确定的各物理性质示出在表3中。关于调色剂颗粒a1至a13,生产条件和通过上述方法确定的各物理性质示出在表4中。
[0369]
[表3]
[0370]
脱模剂种类分子量熔点(℃)sp值((cal/cm3)
1/2
)b1山萮酸山萮酯649758.59b2乙二醇二月桂酸酯426558.97b3乙二醇二硬脂酸酯594728.85b4聚乙烯1194948.45b5费-托蜡283778.11b6巴西棕榈蜡675838.57b7二季戊四醇六月桂酸酯1348789.14
[0371]
[表4]
[0372][0373]
《调色剂1的生产例》
[0374]
使用fm混合器(商品名:fm-10b,由nippon coke&engineering co.,ltd.制造)在转速为3,500rpm的条件下将调色剂颗粒a1(100份)和疏水性二氧化硅颗粒(1份)混合5分钟。在疏水性二氧化硅颗粒的生产中,将3-氨基丙基三乙氧基硅烷和二甲基硅油用作疏水化处理剂。
[0375]
接下来,将2.0份氧化铁颗粒c1注入fm混合器中,并且在转速为3,000rpm的条件下进行混合5分钟,由此获得调色剂混合物。
[0376]
其后,使用300目的网筛(开口48μm)除去粗颗粒以获得调色剂1。
[0377]
《调色剂2至16的生产例》
[0378]
在调色剂1的生产例中,并且如表5所示改变所使用的氧化铁颗粒的种类。除了上述以外,通过与调色剂1的生产例中相同的设备并且在相同的条件下获得调色剂2至16。
[0379]
《调色剂17至23的生产例》
[0380]
在调色剂1的生产例中,并且如表5所示改变使用的调色剂颗粒和氧化铁颗粒的种类。除了上述以外,通过与调色剂1的生产例中相同的设备并且在相同的条件下获得调色剂17至23。
[0381]
《调色剂24至29的生产例》
[0382]
在调色剂1的生产例中,将使用的调色剂颗粒的种类改变为调色剂颗粒a5,并且将使用的氧化铁颗粒的种类改变为氧化铁颗粒c13,并且如表5所示改变氧化铁颗粒的使用量。除了上述以外,通过与调色剂1的生产例中相同的设备并且在相同的条件下获得调色剂24至29。
[0383]
《调色剂30的生产例》
[0384]
使用fm混合器(商品名:fm-10b,由nippon coke&engineering co.,ltd.制造)在转速为3,500rpm的条件下将调色剂颗粒a5(100份)和疏水性二氧化硅颗粒(1份)混合5分钟。在疏水性二氧化硅颗粒的生产中,将3-氨基丙基三乙氧基硅烷和二甲基硅油用作疏水化处理剂。
[0385]
接下来,将0.1份氧化铁颗粒c5注入fm混合器中,并且在转速为3,200rpm的条件下
进行混合3分钟,由此获得调色剂混合物。
[0386]
其后,使用300目的网筛(开口48μm)除去粗颗粒以获得调色剂30。
[0387]
《调色剂31至34的生产例》
[0388]
在调色剂30的生产例中,如表5中所示改变当对氧化铁颗粒进行外部添加处理时的条件。除了上述以外,通过与调色剂30的生产例中相同的设备并且在相同的条件下获得调色剂31至34。
[0389]
《调色剂35的生产例》
[0390]
在调色剂34的生产例中,将使用的调色剂颗粒的种类改变为调色剂颗粒a9。除了上述以外,通过与调色剂34的生产例中相同的设备并且在相同的条件下获得调色剂35。
[0391]
《比较例》
[0392]
《调色剂36至42的生产例》
[0393]
在调色剂1的生产例中,并且如表5中所示改变使用的调色剂颗粒和氧化铁颗粒的种类。除了上述以外,通过与调色剂1的生产例中相同的设备并且在相同的条件下获得调色剂36至42。
[0394]
[表5]
[0395][0396]
关于如上所述生产的各调色剂,通过上述方法确定的各物理性质值示出在表6中。
[0397]
[表6]
[0398][0399]
《评价》
[0400]
考虑到高速机器的定影性评价,使用处理速度改装为500mm/秒的hp laserjet enterprise m609dn。另外,通过连接外部电源进行改装,使得改变转印偏压,如下评价定影膜污染、摩擦定影性、和转印缺陷。
[0401]
《评价1:定影膜污染的评价》
[0402]
通过在常湿常温环境下连续输出50张实心黑色图像之后立即输出3张实心白色图像来评价定影膜污染,并且确定实心白色图像的污染程度。
[0403]
当将例如实心黑色图像等高打印率图像定影时,无法从定影膜释放的调色剂的一部分附着至定影膜。当立即打印实心白色图像时,其后,将残留在定影膜上的调色剂转印到纸上,并且作为纸上的污染物而变得明显。
[0404]
用光学显微镜观察如上所述获得的实心白色图像,并且基于以下标准来评价。评价结果示出在表6中。
[0405]
a:未观察到污染。
[0406]
b:仅观察到污染的点。
[0407]
c:观察到两个以上轻微污染的点。
[0408]
d:观察到污染,但是在整个表面上观察到轻微污染,或者观察到立即可见的清晰的污染。
[0409]
《评价2:摩擦定影性的评价》
[0410]
通过在常湿常温环境下输出实心黑色图像并且测量用擦子摩擦前后的浓度降低率来评价摩擦定影性。
[0411]
使用擦子(商品名:mono,由tombow pencil co.,ltd.制造)以300g的负荷测试定影图像的耐摩擦性。测量用擦子将实心图像往复摩擦10次前后的浓度降低率,并且基于以下标准来评价摩擦定影性。降低率越低,摩擦定影性越好。评价结果示出在表6中。
[0412]
a:浓度降低率为0至3.0%。
[0413]
b:浓度降低率为3.1至10.0%。
[0414]
c:浓度降低率为10.1至15.0%。
[0415]
d:浓度降低率为15.1%以上。
[0416]
《评价3:转印缺陷的评价》
[0417]
通常,当转印偏压高时,容易发生放电,并且可以严格地评价转印缺陷。
[0418]
另外,通常,在使用放置在高湿环境下的厚纸的情况下,转印性严格。
[0419]
使用厚纸(95g/m2,由canon inc.制造),在高温高湿环境(32.5℃/80%rh)下,在通常的转印偏压(0.5kv)下,以2%的打印率的横线的一张间歇模式进行1,500张图像打印。在打印1,500张图像之后,输出一张实心黑色图像。其后,将转印偏压设定至1.5kv,并且输出实心黑色图像。
[0420]
通过将转印偏压改变至1.5kv,并且目视观察输出的实心黑色图像基于以下标准来评价转印缺陷。评价结果示出在表6中。
[0421]
a:未观察到转印缺陷。
[0422]
b:部分观察到浓度不均匀。
[0423]
c:在整个表面上观察到浓度不均匀。
[0424]
d:在黑色实心图像上观察到白色空白部分。
[0425]
[表7]
[0426][0427]
虽然已参照示例性实施方案描述了本发明,但应理解本发明不限于公开的示例性实施方案。所附权利要求的范围应符合最宽泛的解释以涵盖全部此类修改以及等同的结构和功能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1