一种具有核壳结构CoFe2O4@MCM‑48磁性介孔材料的制备方法与流程
本发明磁性复合材料技术领域,具体涉及一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法。
背景技术:
cofe2o4颗粒具有很多种优良的物理化学性能,如电阻率高、矫顽力高、饱和磁化强度适中、磁谱特性好等特性,而活跃在光学、磁学、电学等各领域,是重要的多功能材料。介孔材料mcm-48型分子筛具有独特的结构特点,如比表面积大、孔径均一、三维螺旋面孔道结构、良好的长程周期性和稳定的骨架结构等,而活跃于吸附分离、催化反应、药物释控和半导体材料、电子跃迁光敏剂、碳纤维、非线性光学等主体材料领域。将两种材料相结合可以实现功能互补。
目前人们研究较多的是mcm-41型分子筛包覆cofe2o4尖晶石形成磁性复合材料,而对于mcm-48型分子筛包覆cofe2o4尖晶石报道很少。但是也有部分学者利用溶胶凝胶法分三步制备出cofe2o4@mcm-48复合材料,该复合材料的微观结构为尺寸均匀的球形形貌,比表面积为1100~1300m2/g,孔容为0.85~1.20cm3/g,孔径为2.30~2.90nm,粒径分布范围在200~400nm之间。将该材料应用于铀酰离子的吸附分离,结果表明,初始ph值对铀酰离子的吸附影响最大,最大吸附效率为91.16%,吸附容量为217.97ml/g。另外,通过自助装技术制备出cofe2o4@mcm-48复合材料,作者研究了该材料对水中五种磺胺类抗生素的吸附能力,结果表明15℃时平衡吸附量在68.9μg·g-1(磺胺二甲嘧啶)至99.6μg·g-1(磺胺甲二唑)之间。
目前水热合成法是磁性复合材料cofe2o4@mcm-48分子筛的主要方法,但是水热法制备周期长,消耗能量高,导致目前磁性复合材料cofe2o4@mcm-48分子筛制备成本高,并且磁性复合材料cofe2o4@mcm-48制备过程非常繁琐,合成条件非常苛刻,合成时间较长。
现有技术中存在以下问题:(1)磁性复合材料cofe2o4@mcm-48制备过程非常繁琐,合成条件非常苛刻,合成时间较长,因此简化制备流程是本文想要解决的技术问题;(2)水热合成法是目前磁性复合材料cofe2o4@mcm-48分子筛的主要方法,水热法制备周期长,模板剂消耗量较大,消耗能量高,成本高。这些缺点在一定程度上限制了磁性介孔复合材料的广泛应用。
技术实现要素:
为了解决现有技术中存在的不足,本发明提供的一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,操作简单、条件温和、合成时间短且成本低。
本发明提供的一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,包括以下步骤:
s1,取cofe2o4溶于氯仿中,超声10~20min,得到混合溶液;
s2,取s1中得到的混合溶液加到含十六烷基三甲基溴化铵的水溶液中,继续超声搅拌3~8min,将溶液温度升到50~70℃,然后再持续蒸发1~3min,得到了铁氧体颗粒溶液;s2中使用的十六烷基三甲基溴化铵与s1中使用的cofe2o4的比例为0.0005mol:0.1g;
s3,取s2得到的铁氧体颗粒溶液加入到十六烷基三甲基溴化铵-乙二醇-水溶液中,搅拌5~15min;然后加入氨水调节ph至11,再加入无水乙醇,继续搅拌;接着在搅拌条件下加入正硅酸乙酯,继续搅拌,充分反应后得到复合材料溶液;s3中使用的十六烷基三甲基溴化铵与s1中使用的cofe2o4的比例为0.0007~0.0091mol:0.1g;使用的正硅酸乙酯与s2中使用的cofe2o4的比例为0.006mol:0.1g;十六烷基三甲基溴化铵-乙二醇-水溶液中乙二醇与十六烷基三甲基溴化铵的比例为0~1.3152g:0.0007~0.0091mol;使用的无水乙醇与s1中使用的cofe2o4的比例为30ml:0.1g;
s4,s3中得到的复合材料溶液经过滤、洗涤、干燥,并于马弗炉中450~650℃焙烧3~8h,最终得到具有核壳结构的cofe2o4@mcm-48磁性介孔材料。
优选的,上述的具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,所述cofe2o4与氯仿的比例为0.1~0.3g:3ml。
优选的,上述的具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,s2中,十六烷基三甲基溴化铵的水溶液中十六烷基三甲基溴化铵的浓度为0.8g/100ml。
优选的,上述的具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,s2中,溶液温度为60℃。
优选的,上述的具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,s3中,十六烷基三甲基溴化铵-乙二醇-水溶液的具体配方为每62g水中含有0~1.3152g乙二醇和0.0007~0.0091mol十六烷基三甲基溴化铵,余量为水。
优选的,上述的具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,s4中,于马弗炉中550℃焙烧5h。
与现有技术相比,本发明的制备方法具有以下有益效果:
(1)本发明将铁氧体cofe2o4颗粒和介孔分子筛mcm-48相结合实现了铁氧体和分子筛的功能互补;表面活性剂在水中会自组装成有序结构,同时自组装过程与温度、浓度、添加剂等因素有关,并且表面活性剂通过自组装过程生成的结构对mcm-48型分子筛合成有很强的指导性;硅源与模板剂之间的相互作用过程与介质的ph值、催化剂、有机添加剂、反应条件有关,很大程度上为动力学控制过程;模板剂和硅源物种之间的相互作用是能否形成稳定的杂化介观结构的关键,只有合适类型和强度的相互作用才能促使具有介观结构的固体形成。
利用本发明的方法制备的磁性介孔材料具有特殊的理化性质,诸如大的比表面积、独特的磁性特征、低毒性及表面易于修饰等,使其在生物医学(药物/基因/rna输送)、吸附分离、催化、显像成像等领域有广泛应用。马弗炉中的焙烧时间最短为3h,通过对磁性介孔材料进行表面修饰等进一步的改进,可以应用于更广阔的领域。本发明的方法成本低、毒性低、操作简便、条件温和、耗时短,制备的材料既具有磁性又具有分子筛的介孔孔道,在吸附分离、药物载体方面有巨大的前景。
(2)结构物相方面的改进:本文研究了改变铁酸钴的用量、teos和ctab的比值、乙二醇的用量对复合材料cofe2o4/mcm-48合成的影响,结果表明当铁酸钴的掺入量为0.1g、0.2g、0.3g均能制备出磁性材料cofe2o4@mcm-48,但是在铁酸钴的掺入量为0.2g时制备样品的有序性较高,再增加掺入量样品的有序性就会下降,这是由于在二氧化硅球心的位置加入了铁酸钴磁性纳米颗粒,降低了介孔材料的有序孔道结构;通过调节teos和ctab的摩尔比,发现随着teos/ctab的增加样品的有序性慢慢变高,mcm-48分子筛的物相慢慢显现,且teos/ctab摩尔比的改变对复合材料的磁性有一定的调节作用。
通过调节乙二醇的使用量,我们发现增加乙二醇的使用量可以使样品晶胞体积变大,粒径也随之变大。
(3)磁性方面的改进:通过调节乙二醇的用量,制备出的磁性材料样品饱和磁化强度随着乙二醇使用量不断增加而减少,当乙二醇的使用量由0.328g变为0.658g时样品的饱和磁化强度发生大幅度下降;当乙二醇的使用量为0g时样品的饱和磁化强度最大为0.0450emu/g,这是由于乙二醇使用量的增加导致了样品中硅羟基含量增加,分子筛晶胞体积变大,包覆在铁氧体外层的分子筛厚度变大。
(4)比表面积、孔径、孔容等性能方面的改进:该样品比表面积分布在982.86~1078.46m2/g,孔容分布在:0.69~1.30cm3/g,孔径分布在:1.86~2.52nm。氮吸附测试我们发现当铁酸钴的掺入量为0.2g时,样品的比表面积、孔体积、孔径达到最佳。当乙二醇使用量为1.3152g时样品的比表面积大幅度增加为1619.45m2/g。
(5)形貌方面的改进:图3(a)在垂直于孔轴方向,可以观察到样品孔道的长程有序,显示出直的平行孔,空间互补相通,孔道均匀;图3(b)在平行于孔轴方向,可以观察到样品孔径大小大概在2~3nm左右,这与bjh法测量的孔径大小基本一致;由图3(c)可以清晰的看出复合材料cofe2o4@mcm-48的颗粒形状规整,大小均匀,晶粒尺寸分布在240~280nm之间,没有发生明显的团聚,分散性良好。由图3可以看出中间颜色相较周边较深,磁核cofe2o4(深色区域)外面均匀包裹着mcm-48分子筛(浅色区域),厚度约为20~40nm,采用mcm-48分子筛包裹磁性颗粒可有效增强其化学稳定性,防止团聚,并使材料同时具备分子筛外壳的吸附性能和铁氧体内核的磁学特性。
附图说明
图1为掺入不同量cofe2o4制备的样品大角度xrd图谱;
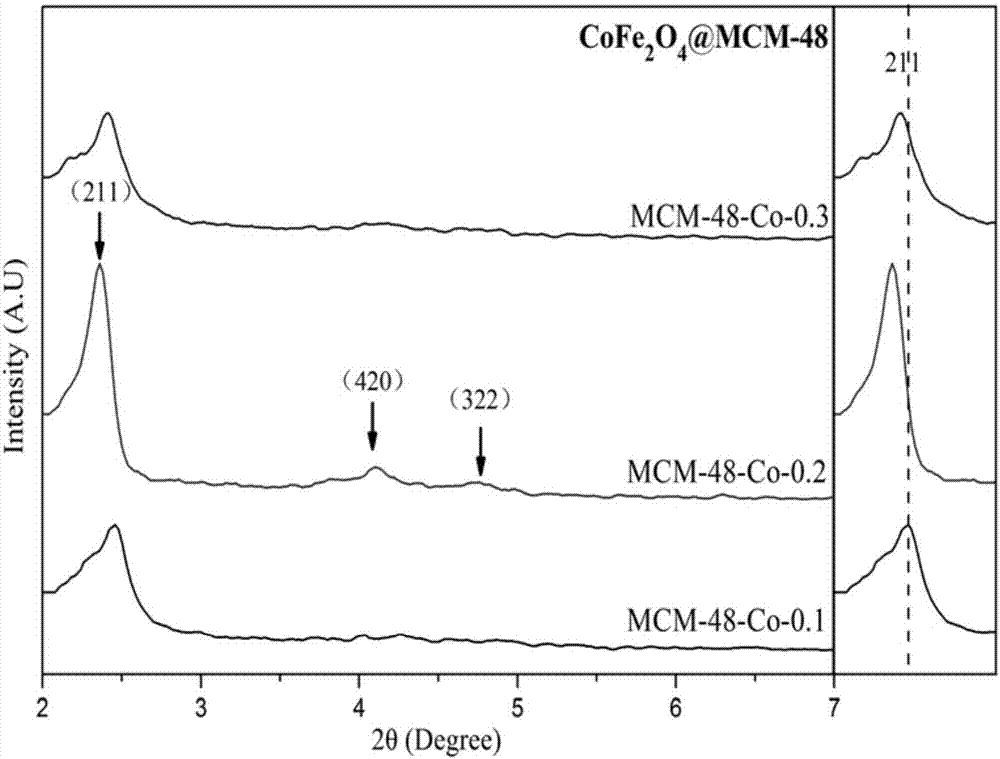
图2为掺杂不同量cofe2o4制备的样品小角度xrd图谱;
图2中左侧图为xrd图谱,右侧图为211衍射峰的偏移情况;
图3为掺入0.2g的cofe2o4时制备cofe2o4@mcm-48磁性介孔材料的透射电镜图;
其中,图3(a)为在垂直于孔轴方向的透射电镜图,图3(b)为在平行于孔轴方向的透射电镜图,图3(c)为在平行于孔轴方向的透射电镜图;
图4是纯相mcm-48型分子筛的电镜图;
图5为掺入不同量cofe2o4制备的样品等温线及孔径分布图;
其中,图5(a)、图5(c)、图5(e)分别为cofe2o4的掺杂量为0.1g、0.2g、0.3g时的等温线,图5(b)、图5(d)、图5(f)分别为cofe2o4的掺杂量为0.1g、0.2g、0.3g时制备的样品的孔径分布;
图6为不同量乙二醇制备的样品大角度xrd图谱;
图7为不同量乙二醇制备的样品小角度xrd图谱;
图7中左侧图为xrd图谱,右侧图为211衍射峰的偏移情况;
图8为不同量乙二醇制备的样品sem图;
图8a、8b、8c时乙二醇添加量1.3152g、0.658g、0g时制备的样品sem图;
图9为不同量乙二醇制备的样品颗粒统计直方图;
图9a、9b、9c时乙二醇添加量1.3152g、0.658g、0g时制备的样品颗粒统计直方图;
图10为不同量乙二醇制备的样品等温线及孔径分布图;
其中,图10(a)、图10(c)、图10(e)、图10(g)分别为乙二醇的掺杂量为0g、0.328g、0.658g、0.986g时的等温线,图10(b)、图11(d)、图10(f)、图10(h)分别为乙二醇的掺杂量为0g、0.328g、0.658g、0.986g时制备的样品的孔径分布;
图11为使用不同量乙二醇制备cofe2o4@mcm-48磁性介孔材料的磁滞回线。
具体实施方式
下面对发明的具体实施方式进行详细描述,但应当理解本发明的保护范围并不受具体实施方式的限制。下列实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。
本发明提供的一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,包括以下步骤:
s1,取cofe2o4溶于氯仿中,超声10~20min,得到混合溶液;
s2,取s1中得到的混合溶液加到含十六烷基三甲基溴化铵的水溶液中,继续超声搅拌3~8min,将溶液温度升到50~70℃,然后再持续蒸发1~3min,得到了铁氧体颗粒溶液;s2中使用的十六烷基三甲基溴化铵与s1中使用的cofe2o4的比例为0.0005mol:0.1g;
s3,取s2得到的铁氧体颗粒溶液加入到十六烷基三甲基溴化铵-乙二醇-水溶液中,搅拌5~15min;然后加入氨水调节ph至11,再加入无水乙醇,继续搅拌;接着在搅拌条件下加入正硅酸乙酯,继续搅拌,充分反应后得到复合材料溶液;s3中是使用的十六烷基三甲基溴化铵与s1中使用的cofe2o4的比例为0.0007~0.0091mol:0.1g;使用的正硅酸乙酯与s2中使用的cofe2o4的比例为0.006mol:0.1g;十六烷基三甲基溴化铵-乙二醇-水溶液中乙二醇与十六烷基三甲基溴化铵的比例为0~1.3152g:0.0007~0.0091mol;使用的无水乙醇与s1中使用的cofe2o4的比例为30ml:0.1g;
s4,s3中得到的复合材料溶液经过滤、洗涤、干燥,并于马弗炉中450~650℃焙烧3~8h,最终得到具有核壳结构的cofe2o4@mcm-48磁性介孔材料。
具体包括以下实施例。
实施例1
一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,具体步骤如下:
s1,取0.2g铁氧体cofe2o4溶于3ml的氯仿中,400w超声15min,得到混合溶液;此处采用的是cofe2o4纳米颗粒;
s2,取s1中得到的一半体积的混合溶液加到25ml的0.8g/100ml十六烷基三甲基溴化铵(ctab)水溶液(即加入了0.0005molctab)中,继续400w超声搅拌5min,将溶液温度升到60℃,然后再持续蒸发2min,以去除有机氯仿,此时得到了表面活性剂稳定的铁氧体颗粒溶液,且铁氧体颗粒表现为亲水性;
s3,取s2得到的铁氧体颗粒溶液加入到62g十六烷基三甲基溴化铵-乙二醇-水溶液中,搅拌10min;然后加入氨水调节ph至11,再加入30ml无水乙醇,继续搅拌5min;接着在高速搅拌条件下加入0.006mol正硅酸乙酯(teos),继续搅拌24h充分反应,充分反应后得到复合材料溶液;其中氨水的浓度为25~28g/100g;
其中,十六烷基三甲基溴化铵-乙二醇-水溶液的具体配方为每62g水中含有0.658g乙二醇和0.0007molctab,余量为水;
s4,s3中得到的复合材料溶液经过滤、洗涤、干燥,并于马弗炉中550℃焙烧5h,以去除模板剂,得到具有核壳结构cofe2o4@mcm-48磁性介孔材料。
实施例2
一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,具体步骤与实施例1相同,区别仅在于s1中使用的cofe2o4的质量为0.1g。
实施例3
一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,具体步骤与实施例1相同,区别仅在于s1中使用的cofe2o4的质量为0.3g。
实施例4
一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,具体步骤与实施例1相同,区别仅在于s3中十六烷基三甲基溴化铵-乙二醇-水溶液的具体配方为每62g水中含有1.3152g乙二醇和0.0007molctab,余量为水。
实施例5
一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,具体步骤与实施例1相同,区别仅在于s3中十六烷基三甲基溴化铵-乙二醇-水溶液的具体配方为每62g水中含有0.986g乙二醇和0.0007molctab,余量为水。
实施例6
一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,具体步骤与实施例1相同,区别仅在于s3中十六烷基三甲基溴化铵-乙二醇-水溶液的具体配方为每62g水中含有0.328g乙二醇和0.0007molctab,余量为水。
实施例7
一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,具体步骤与实施例1相同,区别仅在于s3中十六烷基三甲基溴化铵-乙二醇-水溶液的具体配方为每62g水中含有0g乙二醇和0.0007molctab,余量为水。
实施例8
一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,具体步骤与实施例1相同,区别仅在于s4中马弗炉中温度为450℃,焙烧时间为8h。
实施例9
一种具有核壳结构cofe2o4@mcm-48磁性介孔材料的制备方法,具体步骤与实施例1相同,区别仅在于s4中马弗炉中温度为650℃,焙烧时间为3h。一、我们以实施例1~3制备的材料为例,不同铁酸钴(cofe2o4)用量对cofe2o4@mcm-48磁性介孔材料的影响
我们研究了不同cofe2o4用量对cofe2o4@mcm-48磁性介孔材料制备的影响,制备了mcm-48-co-0.1(实施例2)、mcm-48-co-0.2(实施例1)、mcm-48-co-0.3(实施例3)三个样品,不同cofe2o4掺入量制备样品的参数如表1所示。图1为掺入不同量cofe2o4制备的样品大角度xrd图谱,从图1中我们可以看到有属于220、311、400等晶面的衍射峰存在,对比标准卡片这与jcpds卡库的22-1086(cofe2o4)标准图谱相一致,在2θ=23°时有一个较宽的衍射峰,这是无定型二氧化硅的衍射峰,说明每个材料中均有cofe2o4磁性材料的成分,也有二氧化硅存在,且随着cofe2o4的掺入量增加311峰强度不断增加,cofe2o4的物相变得明显,故我们可以初步判断我们制备的复合材料可能具有核壳结构。
表1不同cofe2o4掺入量制备样品的参数
图2为掺杂不同量cofe2o4制备的样品小角度xrd图谱,图2中左侧图为xrd图谱,右侧图为211衍射峰的偏移情况;从图2左侧图中可以看到三个样品的xrd图谱均有三个相同的衍射峰,根据布拉格定律,它们分别属于211、420和322晶面的衍射。说明三种样品中均含有mcm-48型分子筛,还可以看出当cofe2o4掺入量为0.2g时,制备的样品211衍射峰比其它两个样品211衍射峰高,这说明了当cofe2o4掺入量为0.2g时,复合材料的结晶度和有序性较高,故下述实验中选择cofe2o4的掺入量均为0.2g。
图2右侧图为211衍射峰的偏移情况,其中虚线表示主衍射峰的偏移趋势,可以看到:随着cofe2o4掺杂量的增加,样品的主衍射峰先向低角度偏移,再向高角度偏移,说明其晶胞体积先增大后减小。图3为掺入0.2g的cofe2o4时制备cofe2o4@mcm-48磁性介孔材料的透射电镜图,图3(a)为在垂直于孔轴方向的透射电镜图,可以观察到样品孔道的长程有序,显示出直的平行孔,空间互补相通,孔道均匀;图3(b)为在平行于孔轴方向的透射电镜图,可以观察到样品孔径大小大概在2~3nm左右,这与bjh法测量的孔径大小基本一致,由图3(c)为可以清晰的看出cofe2o4@mcm-48材料的颗粒形状图,可以看出颗粒形状cofe2o4@mcm-48的规整,大小均匀,晶粒尺寸分布在240~280nm之间,没有发生明显的团聚,分散性良好;中间颜色相较周边较深,磁核cofe2o4(深色区域)外面均匀包裹着mcm-48分子筛(浅色区域),厚度约为20~40nm,相比较mcm-48型分子筛的电镜图(参见图4),图4是纯相mcm-48型分子筛的电镜图,可以发现采用mcm-48分子筛包裹磁性颗粒可有效增强其化学稳定性,防止团聚,并使材料同时具备分子筛外壳的吸附性能和铁氧体内核的磁学特性。
图5为掺入不同量cofe2o4制备的样品等温线及孔径分布图,其中,图5(a)、图5(c)、图5(e)分别为cofe2o4的掺杂量为0.1g、0.2g、0.3g时的等温线,图5(b)、图5(d)、图5(f)分别为cofe2o4的掺杂量为0.1g、0.2g、0.3g时制备的样品的孔径分布。由图5可知样品吸附量随相对压强(p/p0)有以下几个变化阶段。在超低压段,n2吸附量迅速增加这是由于n2分子在样品微孔中发生毛细凝聚现象,在低压段,由于介孔孔道表面缓慢吸附一层n2分子导致吸附曲线趋于平缓上升;在中分压段,由于介孔孔道内毛细管凝聚导致n2吸附量迅速增加;在高分压段,由于n2分子的吸附达到逐渐饱和态,所以n2吸附量缓慢递增,根据等温吸附曲线我们可以判断样品中既有微孔又有介孔。如图5所示,各个样品等温线都有一定的突跃,发生突跃的相对压强越大表明样品的孔径越大,另外,突跃陡峭程度可以用来衡量样品孔径是否均匀,变化幅度大,则显示孔径大小均一,即孔径分布窄,中孔体积大。如图三个样品的突跃位置大致相同,所以孔径也大致相同,其中cofe2o4的掺杂量为0.2g时突跃程度最大,所以cofe2o4的掺杂量为0.2g时孔径分布最均匀。由表2和图5中孔径分布图可知样品孔径较为均一,孔径大小集中在2~3nm之间,也可以验证复合材料cofe2o4@mcm-48为中孔物质,其中当cofe2o4的掺杂量为0.2g时孔径分布最均匀。
综上,实施例1中cofe2o4的用量为0.2g时制备的材料具有最好的磁性和核壳结构,其余cofe2o4的用量下效果一般。
表2不同cofe2o4掺入量制备样品的参数
二、我们以实施例1、4~7制备的复合吸附材料为例,说明不同乙二醇用量对cofe2o4@mcm-48复合吸附材料的影响
我们研究了不同乙二醇用量对cofe2o4@mcm-48磁性介孔材料制备的影响,制备了mcm-48-cf-0(实施例7)、mcm-48-cf-0.328(实施例6)、mcm-48-cf-0.658(实施例1)、mcm-48-cf-0.986(实施例5)、mcm-48-cf-1.315(实施例4)五个样品,不同乙二醇掺入量制备样品的实验设计参数如表3所示。
表3不同乙二醇钴掺入量制备样品的实验设计参数
图6为不同量乙二醇制备的样品大角度xrd图谱,从图6中我们可以看到有属于220、311、400等晶面的衍射峰存在。对比标准卡片这与jcpds卡库的22-1086(cofe2o4)标准图谱相一致,在2θ=23°时有一个宽的衍射峰,这是无定型二氧化硅的衍射峰,说明复合材料中既有铁酸钴磁性材料的成分存在,也有二氧化硅的成分存在。
图7为不同量乙二醇制备的样品小角度xrd图谱,图7中左侧图为xrd图谱,右侧图为211衍射峰的偏移情况;从图6左侧图中我们可以看到五个样品的xrd图谱都有三个特征峰,分别属于211、420和322晶面的衍射。这与现有技术中制备的具有ia3d结构的mcm-48分子筛的xrd图谱相符合,说明五种样品均为mcm-48型分子筛,其中可以看出当乙二醇(f108)的使用量为0.986g时,制备的样品211衍射峰比其它四个样品211衍射峰高,这反映了当乙二醇(f108)的使用量为0.986g时,复合材料的有序性较高。图7右侧图为211衍射峰的偏移情况,其中虚线表示主衍射峰的偏移趋势,可以看到:随着cofe2o4掺杂量的增加,样品的主衍射峰先向低角度偏移,再向高角度偏移,说明其晶胞体积先增大后减小。该结论可通过布拉格方程进行解释,由布拉格方程可以知道其晶面间距d变大,从而引起晶胞体积变大;当衍射峰向右偏移时,其晶胞体积减小,因为乙二醇使用量的增加,增加了样品中的硅羟基的含量,从而导致晶胞体积的变大。
图8为不同量乙二醇制备的样品sem图,图8a、8b、8c分别为乙二醇添加量为1.3152g、0.658g、0g时制备的样品;由图8可以发现样品颗粒呈现规则球状,大小较为一致,未出现团聚,而且随着乙二醇的使用量增加样品的粒径也在增加,这与前面xrd分析的样品晶胞体积变大相一致。
为了得到样品的颗粒分布情况,本实验根据得到的扫描电镜图片,使用统计软件对颗粒进行统计,如图9所示,图9为不同量乙二醇制备的样品颗粒统计直方图;图9a、9b、9c是乙二醇添加量1.3152g、0.658g、0g时制备的样品颗粒统计直方图;从直方图我们可以了解到:三个样品中较多的粒子主要集中在448~483.6nm、438.9~472.2nm和372.5~397.6nm范围内,经统计颗粒的平均颗粒尺寸分别是467.21nm、465.69nm和371.39nm。
图10为不同量乙二醇制备的样品等温线及孔径分布图,其中,图10(a)、图10(c)、图10(e)、图10(g)分别为乙二醇的掺杂量为0、0.328g、0.658g、0.986g、1.3152g时的等温线,图10(b)、图10(d)、图10(f)、图10(h)分别为cofe2o4的掺杂量为0、0.328g、0.658g、0.986g、1.3152g时制备的样品的孔径分布。
由图10可知样品吸附量随相对压强(p/p0)有以下几个变化阶段。在0.1以下的超低压段,n2吸附量迅速增加,这是由于n2分子在样品微孔中发生毛细凝聚现象;相对压强在0.1~0.9段氮吸附量缓慢增加,这是由于吸附分子之间的作用,完全填满孔道需要稍高一点的压力;相对压强在0.9~1.0段时n2吸附量又迅速增加,这是因为样品的孔道内表面吸附完全以后样品外表面开始吸附,这是典型的i型微孔等温吸附曲线。
图10中mcm-48-cf-0、mcm-48-cf-0.328、mcm-48-cf-0.658、mcm-48-cf-0.986、mcm-48-cf-1.3152五个样品的等温线在中压段都有突跃趋势说明样品中有介孔存在,所以样品中既有微孔又有介孔。由表4和图10中孔径分布图可知样品孔径较为均一,孔径大小主要集中在1.5~2.5nm之间。当乙二醇=0.328g时,有少量孔径分布在4nm左右,其中当乙二醇=0.658g时孔径分布最均匀。
表4不同量乙二醇制备的样品结构参数
图11为使用不同量乙二醇制备cofe2o4@mcm-48磁性介孔材料的磁滞回线,所有样品的测试环境均相同,均在室温0.5t的磁场下进行测量。由图11可看出cofe2o4@mcm-48磁性介孔材料具有一定的磁性能。
表5为不同量乙二醇制备的样品磁性参数。当磁场增加到5000oe时,样品磁化强度接近饱和,且饱和磁化强度随着乙二醇使用量的增加不断减少。当乙二醇的使用量由0.328g变为0.658g时,样品的饱和磁化强度发生大幅度下降。当乙二醇的使用量为0g时,样品的饱和磁化强度最大为0.0450emu/g,这是由于乙二醇使用量增加样品中硅羟基含量增加,分子筛晶胞体积变大,包覆在铁氧体外层的分子筛厚度变大。样品的磁性相比较cofe2o4发生了大幅度降低,且矫顽力的变化趋势与饱和磁化强度的变化一致,结合xrd图谱可以判断样品是形成了复合材料。研究表明cofe2o4@mcm-48材料是永磁性铁氧体材料,综上所述,我们制备的cofe2o4@mcm-48材料可用于磁分离处理技术研究领域。
表5为不同量乙二醇制备的样品磁性参数
综上,本发明采用溶胶-凝胶法自组装技术,在室温下制备磁性复合材料cofe2o4@mcm-48,并对其结构、磁性能、吸附性能进行研究。xrd表明在使用模板剂十六烷基三甲基溴化铵(ctab)、硅源正硅酸乙酯(teos)、嵌段共聚物乙二醇、助溶剂乙醇、氨水的共同作用下,利用溶胶-凝胶自组装技术制备成功制备出了颗粒尺寸在350~500nm之间的球形cofe2o4@mcm-48磁性介孔材料。该样品比表面积分布在982.86~1078.46m2/g,孔容分布在0.69~1.30cm3/g,孔径分布在1.86~2.52nm。通过氮气吸附测试我们发现当cofe2o4的掺入量为0.2g时,乙二醇的用量为0.658g或者0.328g时,样品的比表面积、孔体积、孔径达到最佳。通过tem研究发现样品磁核cofe2o4外面均匀包裹着mcm-48分子筛,厚度约为20~40nm,说明采用mcm-48包裹修饰磁性颗粒可有效增强其化学稳定性,防止团聚。通过调节teos和ctab的摩尔比,发现随着teos/ctab的增加,样品的有序性慢慢变高,mcm-48分子筛的物相慢慢显现,且teos/ctab摩尔比的改变对复合材料的磁性有一定的调节作用。
需要说明的是,本发明权利要求书中涉及数值范围时,应理解为每个数值范围的两个端点以及两个端点之间任何一个数值均可选用,由于采用的步骤方法与实施例相同,为了防止赘述,本发明描述了优选实施例及其效果,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 还没有人留言评论。精彩留言会获得点赞!
- 磁性胶体核壳结构γ‑Fe2O3及Fe3O4的制备方法与流程
- 一种核壳结构Au@SiO2负载Pt纳米球的制备方法与流程
- 一种核壳结构Ag@TiO2@Pt纳米复合材料的制造方法与工艺
- 一种基于Ag@In核壳结构的高温钎料及其制备方法与流程
- 一种核壳结构Au@Co(OH)2纳米微球的制备方法与流程
- 一种制备核‑壳结构的高介低损钛酸铜钙基陶瓷的方法与流程
- 一种具有核壳结构的微球复合负极材料及其制备方法和应用与制造工艺
- 一种防潮耐水含橡胶籽油的核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种耐盐雾含茶籽油的核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种含锭子油的高流动性核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种核壳结构Au@SiO2负载Pt纳米球的制备方法与流程
- 一种核壳结构Ag@TiO2@Pt纳米复合材料的制造方法与工艺
- 一种基于Ag@In核壳结构的高温钎料及其制备方法与流程
- 一种核壳结构Au@Co(OH)2纳米微球的制备方法与流程
- 一种制备核‑壳结构的高介低损钛酸铜钙基陶瓷的方法与流程
- 一种具有核壳结构的微球复合负极材料及其制备方法和应用与制造工艺
- 一种防潮耐水含橡胶籽油的核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种耐盐雾含茶籽油的核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种含锭子油的高流动性核壳结构纳米改性变压器油及其制备方法与制造工艺
- 一种含椰子油的抗氧化核壳结构纳米改性变压器油及其制备方法与制造工艺