一种掺氮石墨烯量子点电化学制备方法与流程
本发明涉及到一种掺氮石墨烯量子点电化学制备方法,具体是指应用电化学剥离石墨烯产生量子点的过程中引入氮掺杂的制备方法及其应用,该掺氮石墨烯量子点可以应用在诸多领域,如荧光检测和作为超级电容器的电极材料等。本发明属于纳米材料制备及应用领域。
背景技术:
石墨烯是一种碳原子以SP2杂化方式形成的六角型呈蜂巢晶格的二维材料,特殊而完美的结构蕴含了丰富而奇特的物理现象,使石墨烯表现出许多优异的力学、热学、光学和电学特性,在传感器、柔性透明导电薄膜、锂离子电池和超级电容器等领域具有广泛的应用前景。
然而相对于完整晶格结构的石墨烯,石墨烯量子点作为零带隙的半导体闯入了人们的视野。首先,尺寸在10nm以下的零维石墨烯量子点相比二维的石墨烯纳米片,具有更明显的量子限域效应和更强的边缘效应。其次,相较于传统的量子点以及有机染料,石墨烯量子点具有一些更诱人的性质,例如低毒性,强水溶性,高突光稳定性等。
近年来,掺杂型石墨烯量子点成为一大研究热点,异质原子掺入石墨烯的晶格,不仅可以有效地引入带隙,而且还可以增加石墨烯的缺陷和局域的反应活性,从而诱发许多特殊的物理化学特性。同时,掺杂实现了石墨烯量子点能级结构的多样化,在不同的激发光照射下可发出更多不同颜色的发射光,具备高效多色发光特性。目前,氮掺杂型石墨烯量子点的制备及其机理的研究较多,其合成方法从原理上大致可以分为“自下而上”和“自上而下”法。
“自下而上”法主要是以有机小分子为前驱体,成核和碳化过程中或随后进行氮掺杂,包括溶剂化学法和微波水热法。中国专利(CN 105197917 A)用葡萄糖和氨水的微波辐照反应法、(CN 104710983 A)用金属离子与乙醇胺配位的水热反应法,形成氮掺杂石墨烯量子点,但此类方法涉及到微波反应器,高温高压的调控等问题,对设备要求高,操作过程复杂,不能大规模生产制备。
“自上而下”法,通过物理或者化学方法将石墨烯片“裁剪”成较小的石墨烯量子点,同时或者随后加入氮源进行氮掺杂,主要包括水热法和电化学法。水热法工艺主要包括三个步骤:首先制备石墨烯片作为碳源;然后在酸性溶液中酸化和氧化得到浓度更高和尺寸更小的衍生石墨烯片;最后将得到的产物在水热条件下去氧化并引入氮源,即可得到氮掺杂石墨烯量子点。中国专利(CN 103113887 A)以碳纤维磨制成粉末作为碳源,经高温煅烧后分散于浓硝酸中,然后高温长时间回流后加入水和氨水进行水热处理。中国专利(CN 104150473 A)利用氧化石墨和甘氨酸,在氩气氛围高温退火制得氮杂石墨烯片后在强酸中超声切割成氧化氮掺杂石墨烯纳米片,然后水热反应生成量子点。水热反应多需要多个步骤,且强酸参与,反应周期长,氮掺杂石墨烯片的制备反应温度高,生产成本高,限制了实际生产中的应用。而电化学法处理碳材料是制备氮掺杂碳烯量子点的一种简便有效的方法,如中国专利(CN 106757138 A)取无水乙二胺加入高氯酸锂,通氮气,用石墨棒做阴阳极,通入直流电源制备的量子点溶液旋干后重新溶解在水中,再用二氯甲烷萃取。该方法需要惰性气体保护,量子点溶解、萃取等步骤都需要有机溶液中进行,这限制了规模化生产。
到目前为止,条件温和的大批量可控制备氮掺杂石墨烯量子点仍是一个没有有效解决的问题。
技术实现要素:
本发明的目的是为了克服上述现有制备工艺存在的缺陷,提供一种易于规模化大批量生产和应用的掺氮石墨烯量子点电化学制备方法,所述制备方法以直流稳压电源为工作电源,以石墨棒(纯度99.999%)为阳极和阴极,在(NH4)2SO4水溶液中,通过调节添加氨水的量或电解时间,采用电化学氧化的方法刻蚀快速剥离出尺寸均一的氮掺杂石墨烯量子点。本发明采用以下技术方案:
一种氮掺杂石墨烯量子点的制备方法,所述方法包括如下步骤:
A.以石墨棒(优选纯度99.999%的石墨棒)为阳极和阴极,在浓度为0.05~0.5mol/L的(NH4)2SO4水溶液中,通过4~10V恒电位电解对电解池进行预处理,使阳极、阴极表面的石墨层膨胀;
B.向步骤A预处理后的电解池中添加氨水,使氨水中的NH3与步骤A中(NH4)2SO4水溶液中的(NH4)2SO4的摩尔比为1~14:1,保持步骤A中工作电压电解60~180min,然后抽滤电解池中的电解液得到淡黄色滤液,将所述淡黄色滤液搅拌加热,以除去过量氨水;
C.将步骤B得到的滤液在截留分子量为3000~12000Da的透析袋中室温下透析至中性,除去多余的盐离子,得到石墨烯量子点分散液,将所述石墨烯量子点分散液冷冻干燥,得到所述氮掺杂石墨烯量子点。
上述制备方法中,步骤A和步骤B电解所使用的工作电源均为直流稳压电源。
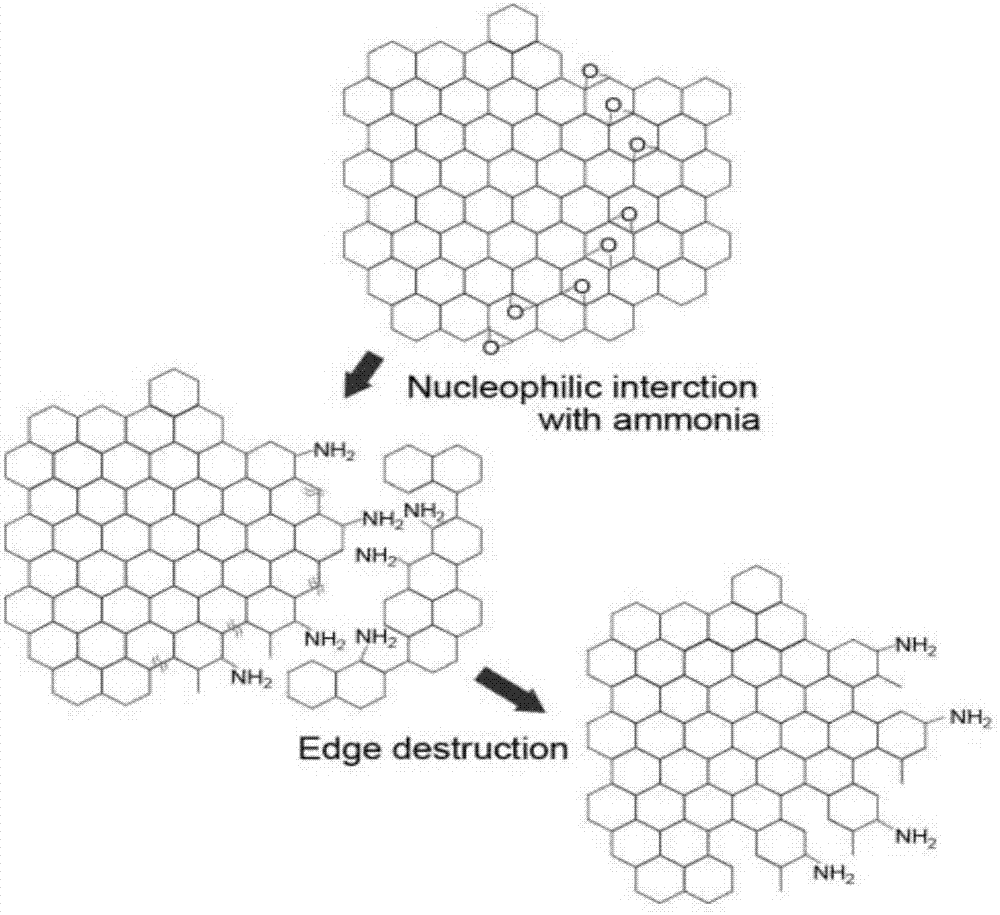
步骤A中,电极工作电压在析氧电位(电解水过程中,理论上O2/H2O的电位是1.23V,但实际上一般需要达到1.36V左右)以上时,能够电解水分子,产生羟基和氧自由基,充当电化学“剪刀”功能,用于切割石墨,产生含氧官能团(电解切割石墨烯量子点机理见图1)。
为了平衡石墨膨胀所需电解时间和效果,本实验最低采用4V工作电压,所述预处理时间为30~120min。硫酸根离子尺寸大于石墨烯层间间距,能够有效的进行插层和剥离。
上述制备方法中,步骤B所述氨水优选浓度为26%,密度约为0.90g/ml的分析纯氨水。
上述制备方法中,步骤B所述抽滤膜采用聚四氟乙烯微孔滤膜,优选孔径为0.22μm,能有效去除溶液中残留物。
上述制备方法中,步骤B所述将该淡黄色滤液搅拌加热优选将该淡黄色滤液搅拌加热40~80℃(氨水的沸点约为36℃);更优选加热至40~80℃条件下,保温1~3小时,来除去过量的氨水。
上述制备方法中,步骤C中所述的透析采用截留分子量为3000~12000Da的透析袋,根据不同的应用需要,选择不同的分子量透析袋;透析时间为48~72小时,直至溶液为中性,可有效去除铵和硫酸根离子。
上述制备方法中,步骤C中,所述冷冻干燥方法为:于温度-84℃,真空度4Pa的条件下冷冻干燥48小时,得到干燥的固态氮掺杂石墨烯量子点粉末。
进一步,所述氮掺杂石墨烯量子点的制备方法优选按如下步骤进行:
A.以纯度为99.999%的石墨棒为阳极和阴极,在浓度为0.05~0.5mol/L的(NH4)2SO4水溶液中,通过4~10V恒电位电解30~120分钟对电解池进行预处理,使阳极、阴极表面的石墨层膨胀;
B.向步骤A预处理后的电解池中添加氨水,使氨水中的NH3与步骤A中(NH4)2SO4水溶液中的(NH4)2SO4的摩尔比为1~14:1,氨水为质量含量在25~28%的分析纯氨水保持步骤A中工作电压电解60~180分钟,然后抽滤电解液得到淡黄色滤液,将所述的淡黄色滤液搅拌加热至40~80℃条件下保温1~3小时,以除去过量氨水;
C.将步骤B得到的淡黄色滤液在截留分子量为3000~12000Da的透析袋中室温下透析48~72小时直至中性,除去多余的盐离子,得到石墨烯量子点分散液,将所述石墨烯量子点分散液于温度-84℃,真空度4Pa的条件下冷冻干燥48小时,得到所述氮掺杂石墨烯量子点。
本发明的有益效果如下:
本发明的电化学制备氮掺杂石墨烯量子点的方法,采用直流电源在弱碱性条件下电解石墨棒,得到水溶性的氮掺杂石墨烯荧光量子点。通过控制氨水添加量,可以调控其荧光颜色,分别得到稳定的蓝、绿荧光量子点,解决了现有方法中需要高温退火、强酸处理、惰性气体保护或有机溶剂体系等苛刻条件,同时设备要求低,易于规模化大批量生产和应用。
所制备的氮掺杂石墨烯量子点,也可应用于电化学储能领域,通过复合该量子点可以提高电极材料活性物质的导电性,同时实现协同作用来提高超级电容器电极材料的性能。
附图说明
图1为本发明制备氮掺杂石墨烯量子点的机理示意图;
图2为实施例1制备的石墨烯量子点的高分辨透射电子显微镜图片;
图3为实施例1制备的石墨烯量子点的X射线衍射图;
图4为实施例1制备的石墨烯量子点的傅里叶变换红外光谱图;
图5为实施例2制备的石墨烯量子点在不同氨水添加量下的荧光照片和光谱图;
图6为实施例2制备的石墨烯量子点的紫外可见吸收光谱图;
图7为氢氧化镍复合实施例3制备的石墨烯量子点前后电极的CV图;
图8为氢氧化镍复合实施例3制备的石墨烯量子点前后电极的GCD图。
具体实施方式
下面结合具体实施例,对本发明所述一种掺氮石墨烯量子点的电化学制备方法这一技术方案及其应用作进一步阐明。以下实施例中氨水为质量含量在26%的分析纯氨水。
实施例1:氮掺杂石墨烯量子点的电化学制备
以石墨棒(纯度99.999%)为阳极和阴极,在50ml的浓度为0.5mol/L(NH4)2SO4水溶液中,通过4V恒电位电解120分钟进行预处理,使电极表面的石墨层膨胀。然后向溶液中逐滴添加10ml氨水,电解180分钟,然后将电解液通过真空抽虑收集得到淡黄色滤液。将滤液在40℃条件下加热搅拌3小时除去多余的氨水。然后在截留分子量为3500Da的透析袋中透析2天时间至中性,除去多余的铵根和硫酸根离子,得到石墨烯量子点水溶液。将得到的透析液冷冻,之后在温度-84℃,真空度4Pa下的冷冻干燥48小时,得到氮掺杂石墨烯量子点固态粉末。
附图2为实施例1制备的石墨烯量子点的高分辨透射电子显微镜图片,如图显示尺寸在2~7nm,分布均匀的石墨烯量子点。
附图3为实施例1制备的石墨烯量子点的X射线衍射图。
附图4为实施例1制备的石墨烯量子点的傅里叶变换红外光谱图。
实施例2:石墨烯量子点的电化学制备及其荧光性能的调控
以石墨棒(纯度99.999%)为阳极和阴极,插入50ml的浓度为0.1mol/L(NH4)2SO4水溶液中,通过8V恒电位电解60分钟进行预处理,使得石墨层膨胀。通过向溶液中分别添加1、2、3、4、5ml氨水,电解90分钟,然后将分散液通过真空抽虑收集得到淡黄色滤液。将滤液在60℃条件下加热搅拌2小时除去多余的氨水。然后在截留分子量为12000Da的透析袋中透析72小时至中性,除去多余的铵根和硫酸根离子,得到石墨烯量子点分散液。然后使用氨水添加量为1、2、3、4、5份的石墨烯量子点分散液分别进行荧光光谱和紫外可见吸收光谱测试。所用测试设备为SHIMADZU RF-6000和UV-2700。
在波长为365nm的紫外光辐射下,1ml氨水添加量的分散液荧光峰分布在450nm,显示出较强的蓝色荧光;作为对比,5ml氨水添加量的分散液荧光峰分布在495nm,显示出绿色荧光。通过荧光光谱测试可知,可以通过调节氨水含量来改变量子点荧光颜色。
电化学法处理使石墨烯量子点表面引入大量的羟基、环氧、羰基和羧基等含氧基团,同时在氨水作用下,分别生成酰胺C-N键和胺C-N键,随着氨水增多,制备的量子点具有更多缺陷,量子点的π-π共轭体系逐渐被破坏,而n-π共轭体系逐渐增多,从而导致荧光颜色改变。
附图5为实施例2制备的石墨烯量子点在不同氨水添加量下的荧光照片和光谱图。
附图6为实施例2制备的石墨烯量子点的紫外可见吸收光谱图。
实施例3:石墨烯量子点的电化学制备及其在电化学储能的应用
以石墨棒(纯度99.999%)为阳极和阴极,插入50ml的浓度为0.05mol/L的(NH4)2SO4水溶液中,通过10V恒电位电解30分钟进行预处理,使得石墨层膨胀。然后向溶液中逐滴添加0.5ml氨水,电解300分钟,然后将分散液通过真空抽虑收集得到淡黄色滤液。将滤液在80℃条件下加热搅拌1小时除去多余的氨水。然后在截留分子量为5000Da的透析袋中透析48小时至中性,除去多余的铵根和硫酸根离子,得到石墨烯量子点分散液。将得到的透析液冷冻,之后在温度-84℃,真空度4Pa下的冷冻干燥48小时,得到氮掺杂石墨烯量子点固态粉末。
氢氧化镍纳米片和石墨烯量子点复合材料制备以及电极组装如下:氢氧化镍纳米片合成:20ml N,N-二甲基甲酰胺+0.2M 2ml醋酸镍溶液,于80℃搅拌1小时,再加20ml水,水热180℃10h。将氢氧化镍纳米片粉末浸渍到含有氮掺杂石墨烯量子点(N-GQDs)的溶液中,超声分散后,真空干燥得到N-GQDs/Ni(OH)2复合材料。
将制备的氢氧化镍复合量子点前后的材料85mg,分别与导电剂炭黑、聚偏氟乙烯粘结剂按照质量比85:10:5混合,然后添加2ml的N-甲基吡咯烷酮后进行研磨均匀,随后分别涂在1cm2的泡沫镍电极上,干燥箱烘干后,连接CHI 760D电化学工作站工作电极,汞/氧化汞作为参比电极,铂片作为对电极,在1mol/L KOH作为电解液的三电极体系中测试这两个电极材料的电化学储能性能并进行对比分析。
附图7为氢氧化镍复合量子点前后的电极材料在20mV/s扫描电压下的循环伏安图对比;图8为氢氧化镍复合量子点的电极材料在1A/g电流密度下的充放电图对比。经计算可知Ni(OH)2@GCD复合材料电极的比容量达到1656F/g,比Ni(OH)2电极(851A/g)高出近两倍,表明氮掺杂石墨烯量子点对于提高电极材料的电容性能具有明显效果。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!