电子晶体化钙铝石型化合物的制备方法与流程
本发明涉及电子晶体化钙铝石型化合物的制备方法。
本申请要求于2016年7月25日申请的日本专利申请2016-145078号的优先权,并将全部内容引用至本文中。
背景技术:
在将cao、al2o3、sio2作为构成成分的铝硅酸钙(calciumaluminosilicate)中存在矿物名称之为“钙铝石(mayenite)”的物质,并且具有与该晶体相同类型的晶体结构的化合物被称之为“钙铝石型化合物”。
据报告,钙铝石型化合物具有12cao·7al2o3(以下,存在缩写为“c12a7”的情况)的代表性组成。上述c12a7的晶体具有如下的独特晶体结构:包含两个分子的晶胞中的66个氧离子中的两个氧离子作为平衡阴离子(counteranion)在由晶体骨架形成并具有正电荷的笼(cage)中的亚纳米空间内包合“游离氧”(o2-)(非专利文献1)。
本发明人们自1980年代开始进行了关于c12a7的研究,相对于通常为绝缘体的钙铝石型化合物,发现了具有导电性的钙铝石型化合物(以下,称之为“导电性钙铝石型化合物”)(专利文献1)。
上述导电性钙铝石型化合物(以下,存在缩写为c12a7:e-的情况)是钙铝石型化合物的上述笼中的游离氧被电子取代的化合物,其电子的理论上最大浓度为2.3×1021cm-3。上述导电性钙铝石型化合物可以称之为无机的电子晶体(electride)型化合物(非专利文献2)。本发明人们所报告的c12a7:e-是在常温、大气中稳定的最初的电子晶体(非专利文献3)。
对上述导电性钙铝石型化合物的制备方法也进行了各种研究。例如提出有,熔化钙铝石型化合物,将其保持在低氧分压的环境气体中,然后进行冷却、凝固而制备上述导电性钙铝石型化合物的方法(专利文献2)、将钙铝石型化合物的前驱体与还原剂混合而进行热处理的方法(专利文献3)。
2007年,通过在钛金属蒸汽中对c12a7单晶、粉末或薄膜进行高温热处理来实现了使更多的电子包含于晶体内的方法,并成功制备出带有金属性的电传导性的c12a7,制备了使用了c12a7:e-的电子释放元件(专利文献4)。
除了上述制备方法之外,还可以列举如下的制备方法:通过ca金属还原法来直接合成钙铝石型化合物的方法(非专利文献4)、不使用ti等还原剂,而是通过放电等离子体(sparkplasma)烧结法来用一步骤合成c12a7:e-的方法(非专利文献5)、通过气相蒸镀法来在基板上进行成膜而形成c12a7电子晶体的薄膜的方法(专利文献5)、或者在钙铝石型化合物的表面配置铝箔并在低氧分压的环境气体下将其保持在高温中而制备导电性钙铝石型化合物(专利文献6)。
(现有技术文献)
(专利文献)
专利文献1:wo2005/000741
专利文献2:wo2005/077859
专利文献3:wo2006/129675
专利文献4:wo2007/060890
专利文献5:wo2013/191210
专利文献6:wo2013/191211
(非专利文献)
非专利文献1:h.b.bartlandt.scheller,neusesjarhrb.mineral.monatsh.12,547-552(1970)。
非专利文献2:s.matsuishi,y.toda,m.miyakawa,k.hayashi,t.kamiya,m.hirano,i.tanaka,andh.hosono,science,301,626-629(2003)。
非专利文献3:s.w.kim,s.matsuishi,m.miyakawa,k.hayashi,m.hirano,andh.hosono,journalaofmaterialsscience:materialsinelectronics,october2007,vol.18,pp5-14。
非专利文献4:s.matsuishi,t.nomura,m.hirano,k.kodama,s.shamoto,andh.hosono,chem.mater.2009,21,2589-2591。
非专利文献5:j.h.chung,j.h.ryu.j.w.eun,b.g.choi,andk.b.shim,electrochem.solid-statelett.2011,vol.14,issue12,e41-e43。
技术实现要素:
(发明所要解决的问题)
如上所述,通过使用还原剂的各种方法来将钙铝石型化合物可转换为导电性钙铝石型化合物、即转换为被电子晶体化的钙铝石型化合物。
在该方法中,如图2所示,在使用了游离氧离子(o2-)和作为还原剂的例如金属ti的情况下,通过以下反应式所示的反应来形成作为还原剂的氧化物的tio2,并从笼中提取游离氧离子并用电子(e-)取代。
ti+2o2-→tio2+4e-···(1)
然而,例如,在专利文献1~4及非专利文献3所示的制备方法中,需要将金属ti、金属钙(ca)等的还原剂与钙铝石型化合物的混合或者在其表面涂敷等的复杂的操作。另外,这些制备方法通常需要1000℃以上的高温反应,甚至需要长时间的加热。
而且,为了最终获得电子晶体,需要在还原反应之后从电子晶体的表面除去作为副产物的tio2、cao等氧化物。因此,很难说它是一种实用性技术,因为它需要大量的时间、能量和复杂的操作。
为了电子晶体的实用化,希求如下的技术:不使用还原剂,并且在更低温且较短时间内以较低能量将钙铝石型化合物电子晶体化的制备技术。
作为不使用还原剂的制备方法,可以列举非专利文献5中所记载的制备方法。然而,该制备方法使用了放电等离子体烧结方法,因此需要等离子体放电装置。另外,在加热至1000℃以上、需要真空设备等的方面,该制备方法与使用还原剂的方法同样地仍然谈不上实用。
本发明是为了解决上述问题而完成的,其目的在于提供一种不需要还原剂,并且在与以往相比低温的反应条件下,以短时间并较少的能量来能够简单地将钙铝石型化合物电子晶体化的制备方法。
(解决问题所采用的措施)
本发明人们深入研究的结果,发现了通过在加热状态下对钙铝石型化合物施加电压并使电流通过来将钙铝石型化合物电子晶体化的方法。
即,本发明的主旨如下。
(1)一种电子晶体化钙铝石型化合物的制备方法,通过在加热状态下对钙铝石型化合物施加电压而使电流直接流动于上述钙铝石型化合物中来使上述钙铝石型化合物电子晶体化。
(2)根据上述技术方案(1)所述的电子晶体化钙铝石型化合物的制备方法,上述钙铝石型化合物的电阻率成为1.0×105ω·cm以下为止施加电压。
(3)根据上述技术方案(2)所述的电子晶体化钙铝石型化合物的制备方法,对上述钙铝石型化合物提供正极和负极而使电流直接流动于上述钙铝石型化合物中。
(4)根据上述技术方案(1)至(3)中任一项所述的电子晶体化钙铝石型化合物的制备方法,上述电子晶体化在惰性气体的环境气体下进行。
(5)根据上述技术方案(1)至(4)中任一项所述的电子晶体化钙铝石型化合物的制备方法,上述电子晶体化钙铝石型化合物的电子浓度为1.0×1020/cm3以上。
(发明的效果)
根据本发明提供一种不需要还原剂,并且在与以往的方法相比低温的反应条件下,以短时间并较少的能量来能够简单地将钙铝石型化合物电子晶体化的制备方法。
此外,通过使电流直接流动于钙铝石型化合物中来能够使上述钙铝石型化合物电子晶体化,因此能够简便地制备出微加工的导电性钙铝石型化合物。
附图说明
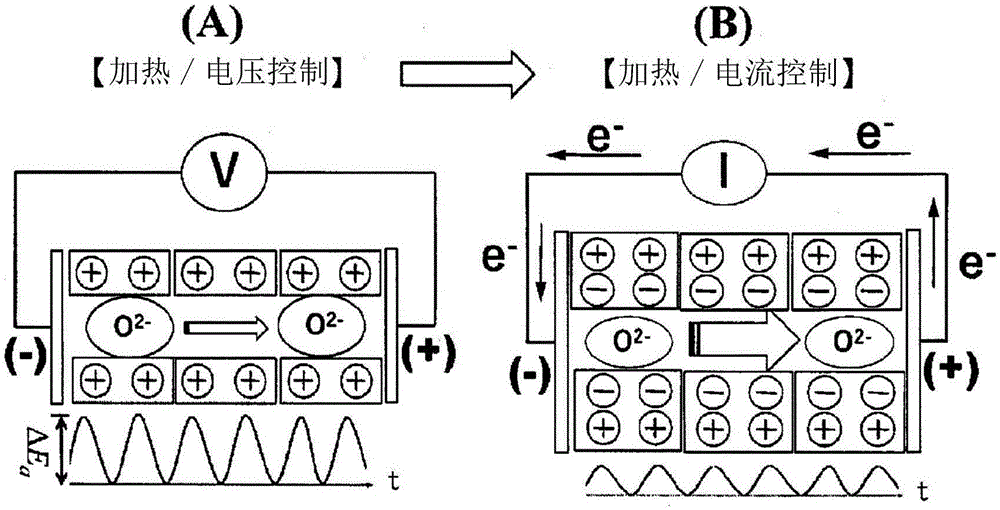
图1为示出本发明的制备方法中的加热/电压控制工序(a)、加热/电流控制工序(b)的概念的模拟图。
图2为示出通过还原法来提取游离氧离子的以往的方法的概念的图。
图3为使用加热炉而制备电子晶体的方法的概念图。
图4为示出在图3所示的实验方法中,比较例1中的试样温度(左纵轴)和输入(in)传感器及输出(out)传感器所测定的氧脱离压力(右纵轴)的时间(横轴)变化的曲线图。
图5为示出在图3所示的实验方法中,比较例2的试样温度(左纵轴)和输入传感器及输出传感器的所测定的氧脱离压力(右纵轴)的时间(横轴)变化的曲线图。
图6为针对区域(area)i(电压控制区域)及区域ii(电流控制区域)分别示出单晶被加热体的加热试验中的试样温度(左纵轴)、施加电压/电流及氧脱离压力(右纵轴)的时间(横轴)变化的曲线图。
图7为示出在区域i中的电压控制下(100v及10v)的被加热体内的电流(纵轴)的变化与加热温度(横轴)之间的关系(i-t曲线)的曲线图。
图8为示出在区域i中的电压控制下(100v及10v)的被加热体的电阻(纵轴)的变化与加热温度(横轴)之间的关系(t-ohm曲线)的曲线图。
图9为示出在区域i中的电压控制下(100v)的被加热体内的电流(纵轴)变化与施加电压(横轴)之间的关系(i-v曲线)的曲线图。
图10为示出在区域i中的电压控制下(10v)的被加热体内的电流(纵轴)变化与施加电压的关系(i-v曲线)的曲线图。
图11为示出在区域ii的电流控制及cd控制下的试样内的电流(左纵轴)变化和施加电压(右纵轴)和加热温度之间的关系(i-v-t曲线)的曲线图。
图12为示出在区域ii的电流控制及cd控制下的试样的电阻(左纵轴)变化和加热温度(横轴)之间的关系(t-ohm曲线)的曲线图。
图13为图12中的电阻的转变温度(925k)部分的放大图。
图14为示出在区域ii的电流控制及cd控制下的氧脱离压力(纵轴)的变化和加热温度(横轴)之间的关系(p-t曲线)的曲线图。
图15为示出在正极侧脱离的单原子氧使正极的钼(mo)氧化的状态的模拟图(b)。
图16为示出将区域ii的电流控制及cd控制下的氧脱离压力比(纵轴)的变化和加热温度(横轴)之间的关系(p-t曲线)与未施加电压的情况进行比较的曲线图。
图17为示出区域ii的电流控制及cd控制下的加热温度(左纵轴)、功耗(右纵轴)及氧脱离压力的时间(横轴)变化(t-w-time曲线)的曲线图。
图18为示出实施例2中的施加电压与电流之间的关系的曲线图。
图19为示出实施例2中的电流控制时的经过时间与电流之间的关系的曲线图。
图20为示出实施例3中的施加电压与电流之间的关系的曲线图。
图21为示出实施例3中的电流控制时的经过时间与电流之间的关系的曲线图。
具体实施方式
以下,详细说明本发明的实施方式。
在本发明中所使用的钙铝石型化合物具有由12cao·7al2o3表示的代表组成。
上述钙铝石型化合物的结晶体是笼状的结构(笼)共享其壁面并三维连接而构成。通常,钙铝石型化合物的笼的内部包含有o2-等的阴离子(anion),但是可以通过化学处理来这些被别的阴离子或传导电子取代。作为上述阴离子虽没有特别限定,但可以列举o2-离子、f-离子、cl-离子等。形成钙铝石型化合物的骨架的上述笼中通常包含有钙(ca)、铝(al)等的阳离子(cation),这些阳离子中的一部分或全部被其他的阳离子取代也可。
作为钙铝石型化合物虽没有特别限定,但具体地可以列举如下的实例。
·仅由ca、al及o原子形成的化合物;
·所有的ca都被其以外的碱土金属离子等的阳离子取代的化合物,例如铝酸锶(sr12al14o33);
·一部分ca被其他的阳离子取代的化合物,例如,作为ca与sr的混合比任意改变的混合晶体的铝酸锶钙(ca12-xsrxal14o33);
·al被其以外的阳离子取代的化合物,例如,作为硅取代型钙铝石型化合物的ca12al10si4o35;或者,
·阳离子和阴离子均被取代的化合物,例如,氯硅铝钙石(wadalite)ca12al10si4o32:6cl-。
在本发明中所使用的钙铝石型化合物的形状虽没有特别限定,但通常可以是粉末状、微粒状、颗粒状、块(bulk)状、由气相法等获得的薄膜状等。此外,使用通过烧结粉末状钙铝石型化合物而获得的烧结体、或者作为块状的一例使用通过区熔(fz,floatingzone)法等而获得的单晶也可。
此外,还可以使用用于制备钙铝石型化合物的原料,并从原料贯穿到导电性钙铝石型化合物的制备。在这种情况下,具体地,在将c12a7作为例子的情况下,使用成为其钙成分和铝成分的原料即可,作为钙成分可以列举碳酸钙、氢氧化钙、氧化钙或金属钙等,作为铝成分可以列举氧化铝、氢氧化铝,金属铝等。这些原料可以使用阳离子比率依照目标物质的化学计量学而混合的物质。此外,还可以使用caal2o4、ca3al2o6等的钙和铝的混合氧化物。
其中,在本发明中,由于氧离子等的笼内的阴离子的移动是顺畅,因此钙铝石型化合物的形状优选为单晶。
本发明的制备方法的特征在于,通过在加热状态下施加电压并使电流直接流动于上述钙铝石型化合物中来使上述钙铝石型化合物电子晶体化。以下,进行具体说明。
在本发明的制备方法中,对钙铝石型化合物施加电压并使电流直接流动。具体地、只要是成为原料的钙铝石型化合物中直接流动电流的话则没有特别限定,但是具体地,给钙铝石型化合物提供成为正极和负极的电极,并从这些电极施加电压而使电流直接流动于钙铝石型化合物中。
上述电极的材料虽没有特别限定,但是通常选择对加热的高温条件具有耐久性的材料、耐氧化的材料,可列举为c(碳)、ti、ni、mo、w、ta、pt、ni-cr合金等,优选为可以通过溅射等的蒸镀法而提供的pt、ti、ni等金属。
上述电极的提供方法虽没有特别限定,但是具体地,存在如下方法:通过机械性接触而设置电极的方法、通过溅射等的蒸镀法而设置的方法。其中,从对任意大小的试样的任意部位设置任意大小(例如,极小的面积)的电极的方面出发,优选的是通过蒸镀法而提供的方法。
作为使电流直接流动于钙铝石型化合物中的方法,通过在导电性的容器中填充钙铝石型化合物并向容器施加电压来使电流直接流动于钙铝石型化合物中。作为导电性的容器的材料,可以列举金属、石墨等,但并没有特别限定。只要是填充在其内部的钙铝石型化合物中直接流动电流的话,上述容器的形状并没有特别限定。
作为具体的方法,可以列举例如,圆柱形的绝缘体容器的两端的盖子使用导电性的材料,在盖子之间填充钙铝石型化合物,并且使电流流动于上述的盖子的方法等。
其中,在操作容易并且能够减少所需的能量的观点来看,优选对钙铝石型化合物提供电极而使电流流动的方法。
在本发明的制备方法中,在对上述的钙铝石型化合物施加电压时,将钙铝石型化合物置于加热状态下。在这里,使钙铝石型化合物的笼内的游离氧离子脱离到笼外并取而代之电子被包合的情况称之为电子晶体化。在这里,被加为了进行电子晶体化而在加热条件下施加电压的钙铝石型化合物称之为被加热体。上述被加热体可以是提供有上述电极的钙铝石型化合物。
加热方法并没有特别限定,但可以是对上述被加热体进行加热器等的外部加热,也可以利用电流流动于上述被加热体中而所产生的热量(以下,存在称之为自我加热的情况)来进行加热。
上述被加热体的加热温度并没有特别限定,可以根据电子晶体化的进程度而适当地选择,但通常是相比所使用的钙铝石型化合物的熔点低的温度,例如在c12a7的情况下,为低于1450℃的温度。越是高温有利于钙铝石型化合物中的游离氧离子的运动性的升高,但是在较低的加热温度有利于能量效率以及不需要高温热源的方面是优选的。此外,在防止被电子晶体化的钙铝石型化合物的氧化的方面,更优选为400℃以下。此外,只要在可获得本发明的效果的范围内,加热的温度履历并没有特别限定,可以连续地进行或间断地进行,也可以以恒定温度进行或者使温度阶段性地上升、下降。
施加于被加热体上的电压并没有特别限定,只要是可以获得本发明的效果即可适当地设定,但是通常大于0v,如后所述,只要是能够施加电压而电流流动于钙铝石型化合物中的话,其电压不会被特别指定。优选的电压的大小依赖于加热温度下的钙铝石型化合物的离子传导率。上限为低于引起介电击穿的电压。考虑到电源装置等的便利性,优选为200v以下。
施加电压的履历并没有特别限定,只要可以获得本发明的效果,可以连续地进行或间断地进行,也可以以恒定电压进行或者使电压阶段性地上升、下降。
只要可以获得本发明的效果,直接流动于上述被加热体中的电流的值并不特别限定。具体地,只要能够确认电流直接流动于上述被加热体中的情况即可,其电流密度通常为0.001acm-2以上且10acm-2以下。
通常,由于钙铝石型化合物是绝缘体,因此电流极难流动。通过后述的具体方法等而对钙铝石型化合物进行加热及电压施加的期间,通常使电流流动。关于使电流流动的具体方法在后叙述。
本发明的制备方法可以适当组合上述的加热方法、上述的电压的施加方法及上述电流的通电方法而制备,并且各方法的具体组合并不特别限定,优选地,例如,可以列举以下的条件1~3。
条件1:将加热温度设为规定的温度,并使电压阶段性地上升的方法
具体地,在对上述被加热体未施加电压的状态下,首先加热到规定的温度,然后将施加于上述被加热体的电压从0v开始阶段性地施加的方法。
条件2:将电压设为规定的电压,并使加热温度阶段性地上升的方法
具体地,设定在常温下施加于上述被加热体的电压,然后使温度从常温阶段性地上升的方法。
条件3:使加热温度和电压同时上升的方法
具体地,从常温及未施加电压的状态一边对上述被加热物体同时调节加热温度及施加电压,一边对上述被加热物体阶段性地进行加热及电压施加。
虽然通过对上述被加热物体进行加热及电压施加来使钙铝石型化合物被电子晶体化,但是被电子晶体化的过程优选的是经过如下步骤。
通常,由于钙铝石型化合物在常温下是绝缘体,所以在刚开始被加施加电压的时点上述被加热体中电流几乎不流动,即使流动也仅流动极微量的电流。继续进行加热及电压施加的话,通常接着观察到流动于上述被加热体中的电流密度的激增的同时所施加的电压的减少。此时的电流密度并没有特别限定,因为通常进行急剧的变化,因此难以特别指定该值,但基准为1acm-2,但通常是0.1acm-2以上。在本说明书中,将观察到上述现象为止的工序称之为加热下的电压控制工序(以下,称之为加热/电压控制工序)。
在上述加热/电压控制工序中,通常,向上述被加热体的电压施加是在施加电压均衡下进行控制被加(以下,称之为电压控制)。这是因为,通常在该工序中,上述被加热体中电流几乎不流动,控制施加电压的话则容易管理制备。
在观察到上述的电流密度增加及施加电压减少的时点(以下,称之为电流密度増加点)之后,与刚开始进行加热及电压施加时相比,认为上述被加热体变成为电流容易流动的状态。
在上述电流密度増加点之后,继续进行向上述被加热体的加热的同时,将电压施加的控制方法从施加电压均衡的控制转移到流动于被加热体的电流值均衡的控制(以下,称之为电流控制)。通过转移到上述电流控制并使电流流动于上述被加热体来使上述被加热体中的氧脱离的同时供应电子。由此,推进钙铝石型化合物的电子晶体化。
通过将电流控制进行至达到所期望的电子浓度为止而可以获得导电性钙铝石型化合物。在本说明书中,将在上述电流密度増加点之后获得所期望的导电性钙铝石型化合物并结束电压的施加为止的工序称之为加热下的电流控制工序(以下,称之为加热/电流控制工序)。
上述加热/电流控制工序是上述被加热体达到所期望的的电子浓度为止适当进行,但通常上述被加热体的电阻率随着电子晶体化的推进而减少,因此继续进行到电阻率充分减少为止。具体的电阻率的值虽没有特别限定,但通常进行至成为1.0×105ω·cm以下为止的情况为基准。
另一方面,上述被加热体的电传导率通常随着电子晶体化的推进而增加。因此,电传导率可以用作电子晶体化的基准,通常继续进行至电传导率成为1.0×10-4s/cm以上为止。
此外,还可以通过观察上述被加热体的颜色来判断。具体地,上述被加热体成为对应于后述的等级(grade)的所期望的颜色为止使电流流动即可。例如,如果期望后述的等级c的导电性钙铝石型化合物的话,将上述加热/电流控制工序继续进行至上述被加热体变成黑色为止即可。
只要是能够获得本发明的效果,从上述加热/电压控制工序转移到上述加热/电流控制工序的方法并没有特别限定,能够适当组合已知方法而进行。具体地,例如,可以列举使用对应于100v/10a等的大功率的电源装置而适当地切换的方法、分别准备在两个工序中所使用的电压施加装置并在刚经过上述电流密度増加点进行切换的方法等。
在本发明制备方法中,能够在没有特别限定压力条件下进行制备,可以在大气压下、减压条件下以及加压条件等的任一条件下制备。
在本发明的制备方法中,能够在没有特别限定周围的环境气体的条件下进行制备。优选地,氧气分压越低可促进从钙铝石型化合物的游离氧离子的脱离,因此,优选地,将被加热物体保持在100pa以下的低氧气分压的环境气体中、尤其是作为氧气分压极少的环境气体的极低氧气分压的环境气体中。上述极低氧气分压的环境气体具体是指,在惰性气体的环境气体下,将氧气分压降低到10-15pa(10-20atom)以下左右的环境气体、或者真空度为10-5pa以下的状态。惰性气体并没有特别限定,可以使用氮气,氩气等,优选为氩气。
尤其,如上所述,在加热温度超过400℃的情况下,氧气分压高的话,被电子晶体化的钙铝石型化合物变得容易被氧化,因此优选为极低氧气分压的环境气体,加热温度为400℃以下的话,则对环境气体并没有特别限定。
根据本发明的制备方法,随着电子晶体化的推进,钙铝石型化合物中的游离氧离子脱离。如上所述,氧气分压对电子晶体化及被电子晶体化的钙铝石型化合物产生影响,因此优选为一边除去上述脱离的氧气一边进行制备。
以每单位体积的电子浓度作为基准进行分类的话,导电性钙铝石型化合物可分类为等级a到等级c的以下三个等级。
[表1]
通过本发明的制备方法获得的导电性钙铝石型化合物可以通过上述三个等级中的任一等级来制备,但最适合为电子浓度最高且每单位体积的电子释放能力高的上述等级c的制造。
具体地,电子浓度的理论上的最大值为2.3×1021cm-3,因此可以获得电子浓度为1.0×1020/cm3以上且2.3×1021cm-3以下的导电性钙铝石型化合物。
此外,所获得的导电性钙铝石型化合物其电子密度可以是均匀的也可以是不均匀的,但优选为均匀的。此外,不均匀的导电性钙铝石型化合物的上述等级判断是由目视(肉眼观察)的颜色来判断,最高的电子密度的测定值用作电子浓度。
此外,通过本发明的制备方法获得的导电性钙铝石型化合物在室温下的电传导率在理论上的最大值为1500s/cm左右,因此优选为1.0s/cm以上,更优选为100s/cm以上且优选为1500s/cm以下。
将参照附图更详细地描述本发明的制备方法中的上述各工序。图1是示出加热/电压控制工序(a)、加热/电流控制工序(b)的概念的模拟图。如图1的(a)所示,在上述加热/电压控制工序中,进行加热而使钙铝石型化合物的笼中的游离氧离子(o2-)活化,从而使离子传导性变高而实现氧离子传导体化。
接下来,如图1的(b)所示,在被加热体的电阻下降之后转换到加热下的电流控制而使带电电子在电压施加电路中流动。在本说明书中,将为了上述能障(energybarrier)(δe)抑制为低或缓解而强制使电流向电势负载方向流动的情况称之为电流驱动(以下,称之为cd控制)。在cd控制下使笼空间保持电中性状态并使势垒(potentialbarrier)下降,据此使氧离子的移动变得容易。
通过上述各工序将电子剥离而从原子极化状态达到电子极化状态,电子极化后的电子原样残留在笼中,氧原子欲要从钙铝石型化合物脱离,但此时的氧脱离压力在加热作用下上升。“氧脱离”是指,通过多个工序而游离氧离子从笼内离开的现象。本说明书中的“氧脱离压力”是在ar等的惰性气体的环境气体中的o2分压。
图3为示出使用加热炉而制备电子晶体的方法的一形态的概念图。该例子为使用加热炉的形态,但加热可以是加热器等的外部加热或内部电阻加热。将c12a7:o2-作为被加热体而装入于极低氧气分压的ar流动的石英管1中。加热炉3上安装极低氧分压控制装置4。极低氧分压控制装置4由含有氧的气体的入口侧氧传感器41、氧气泵42、气体的出口侧氧传感器43、循环泵44构成,极低氧气分压气体供给到加热炉3内。加热环境气体是流入到石英管1内的极低氧气分压的氩气环境气体。在加热下的电压控制及加热下的电流控制的作用下o2从放置在该环境气体下的被加热体2脱离,含有所脱离的氧气的氩气气体流出至石英管1外,并循环到极低氧气分压控制装置4。
在上述加热/电压控制工序中,在200v以下的范围内施加电压的话,加热温度越高温,离子传导率越变高(低电阻化),因此在高温下能够以低电压进行电子晶体化。为了尽可能在低温下进行电子晶体化,在低温下离子传导率低的情况下,施加较高的电压的话,则与高电压负载成比例而钙铝石型化合物内的电流增大。然而,高温加热到300℃以上的话,与温度的上升一同电阻减少,因此负载电压下降的同时氧离子传导率上升,并且钙铝石型化合物内的电流上升。因此,电压范围根据与温度的关系控制在0v~200v的范围即可。然而,在这种情况下,如图1的(a)所示,为了扩宽笼之间的空间上的狭窄的瓶颈,氧阴离子的笼之间的移动是超越能障而进行。
本发明的制备方法的反应机理推算如下。
构成钙铝石型化合物的笼的氧原子与钙原子、铝原子牢固结合,除非晶格被破坏,否则不被脱离。另一方面,笼中的游离氧离子松散地结合。
如果给钙铝石型化合物施加电压的话,笼虽然不能够移动,但氧离子向电流的流动方向、具体地被正极(+)牵引而被笼壁的氧离子取代并进行移动。此时,需要施加超过取代由ca-al-o构成的钙铝石型化合物的笼壁的氧离子所需的势垒(δe)的电压。于是,被加热体的电阻急剧下降且电流密度急剧增加为止,使加热温度和/或电压阶段性地上升。另外,此时的离子传导性通过电阻的测量而被求出。
正极附近的钙铝石型化合物被电解成导电性钙铝石型化合物和氧,其结果氧被释放到笼外。通过因加热而游离氧离子的离子电传导性上升来移动到正极附近的游离氧逐一被电子取代,从而推算为电子晶体化在推进的情况。
因此,以往是使用还原剂而使游离氧离子脱离,但本发明能够在不使用还原剂的情况下使游离氧离子脱离而实现电子晶体化。
实施例
以下,将根据实施例详细说明本发明的制备方法。
(实施例1)
[加热下的电压控制和加热下的电流控制]
将由fz法获得的c12a7单晶体切成10mm×5mm×1mm的大小,如图1所示,通过溅射在两端(端部面面积:5mm×1mm)蒸镀铂(pt)薄膜,并且将正电极和电极的两电极通过钼(mo)线来安装而用作被加热体。按照如图6所示的履历,在加热条件下对该被加热体施加电压。具体地,在电压控制和电流控制下进行了1000℃为止、及1200℃为止的高温处理。处理环境气体为进行了极低氧气分压控制的ar气(ar流量:3l/分钟、平均流速5.2cm/秒、大气压、常温)。作为极低氧气分压控制装置使用了佳能机械(canonmachinery)(株)公司制的装置。如图3所示,由输出传感器和输入传感器测量出氧分压。关于极低氧气分压控制,下同。
其结果,如图6中的右纵轴所示,在约650℃的稳态温度状态下产生大量的氧脱离,并且再加热至1000℃时观察到氧的再脱离压力上升。在同图中“区域i”所示的0分钟~119分钟的区域中,在电压控制下对被加热体进行了阶段性的升温“(1)~(6)”,在119分钟之后的区域ii中,从电压控制转移到电流控制(cd投入),进而在实验的后半部分中进行了使电流控制值增加的cd控制。
如图7所示,在区域i的电压施加控制中,证实了由于加热温度变化(升温速度10k/min)而试样中的电流密度从0.1a·cm-2迅速上升的情况。
检查此时的上述被加热体的电阻的话,如图8所示,发现了与加热温度上升一同上述被加热体的电阻急剧减少的情况。在图8中以箭头(1)、(2)所示的加热温度之前的状态表现出被加热体的绝缘体行为,并且在施加100v下因加热而电阻逐渐减少而表现出离子电传导体化的行为,在以箭头(2)所示的加热阶段中将负载从100v改变到10v并进一步继续加热的话,在以箭头(4)所示的加热阶段发现了电阻几乎为零且推进了电子晶体化的情况。
[施加电压的影响]
于是,通过增加或减少施加电压来研究了在恒定加热温度下的施加电压和试样内的电流之间的关系(i-v曲线)。其结果,如图9、图10所示,发现了在高温加热状态下电压增加变化和减少变化中出现了滞后现象(hysteresis)的情况。即,这种不可逆的变化意味着游离氧离子在被加热体内空间上偏于一方而存在的情况被加。
在图9的(1)的低温(379.3k)状态下,由于钙铝石型化合物为绝缘状态,因此在电压上升和下降时电流几乎不流动。如果在(2)的温度(432k)下的加热时发现了某种程度的离子传导性的话,则在电压升压时和电压降压时会做出不可逆的动作。即,可知升压时移动到正极的游离氧离子在降压时其一部分不会返回到原来位置并偏于一方而存在的情况。与此相对,阱(trap)是指,在一个方向上负载电压时所发生的现象。
[加热温度的影响]
图11示出了区域ii的电流控制以及cd控制下的被加热体中的电流/电压的温度变化。从电流控制开始(730k)的约18v开始,在加热温度达到约819k的阶段观察到了试样电压的急剧变化,并且在925k下降低至约7v为止的阶段在cd控制下使被加热体内的电流逐渐增加。
如图12、图13(图12的局部放大图)所示,此时的上述被加热体的电阻在925k下进一步急剧减少。于是,在下文中,将作为该电阻的转变温度的925k称之为3t(=触发转变温度(triggeringtransitiontemperature))。同样地,图14示出了氧脱离压力(ar中的o2分圧)与加热温度之间的关系。在3t处观察到了氧脱离压力的急剧上升的同时,在3t之后观察到了随着加热温度的上升而氧脱离压力的上升。图15示出在正极侧脱离到ar环境气体中的氧使安装在正极上的钼线的钼(mo)氧化的状态的模拟图。
图16示出,在cd控制(电压施加10v)的情况下和未安装电极(未施加电压)的被加热体的情况下比较了平衡脱离压力比(pio2/piio2)和加热温度(t)的关系的结果,但是可以看出cd投入中3t温度低温化(cd有效)的情况。pio2和piio2分别是由输入传感器测量的o2分压(pio2)和由输出传感器测量的o2分压(piio2)。
[功耗的评估]
图17示出了cd控制之后的温度、功耗和氧脱离压力的时间变化。功耗成为峰值的是电流控制状态下的3t温度维持状态的后半部分,且在氧脱离压力的峰值之后。之后,虽然功耗暂时减少,但是在cd控制下使被加热体内的电流增加的情况下,它转变为增加而饱和成最大500w/cm2左右。
(实施例2)
将由fz法获得的c12a7单晶体切成10mm×5mm×1mm的大小,通过溅射在两端蒸镀铂薄膜,并将各末端分别作为正电极及负电极。对上述正电极及负电极安装钼引线而用作被加热体。
首先,在大气压下,对被加热体施加了100v的施加电压的结果,在试样温度30℃附近观察到了1pa左右的电流(电流密度20pa·cm-2)。
然后,切换为可施加最大100v的电压以及可流动最大30a的电流的电源。在从常温以10℃单位升温之后,保持上述被加热体的温度,在各温度下100v为止施加电压。该过程重复进行达到180℃为止。用本电源能够检测出的最小电流为0.1ma,但在上述条件下没有观测到电流。
在上述被加热体的温度保持在180℃并施加了99v时观测到了0.6ma的电流(电流密度12ma·cm-2)(图18)。该时点的被加热体的电阻率为1.7×104ω·cm,这是表现出被加热体向等级b以上的电子晶体转变的情况。电压保持在100v的结果,约25秒之后观测到了约550ma的电流值的急剧增加,与此同时,使能够施加100v程度的电流不流动于试样中。
其中,将被加热体的电压施加方法从上述电压控制转换到电流控制(图19)。自成为电流控制后大约100秒之后确认了被加热体的状态的结果,被加热体变成黑色且c12a7被电子晶体化了。在该被加热体中发生了电子浓度的不均匀。最大部分为1.0×1021/cm3左右,最低部分为1.0×1019/cm3左右。上述的被电子晶体化的被加热体(本实施例的电子晶体化钙铝石型化合物)中的电子浓度最高的部分的电传导率为5×102s/cm。
[实施例3]
以与实施例2相同的流程将c12a7的加热温度从常温升温到210℃后,保持在210℃,之后,进行了与实施例1相同的实验的结果,施加了约70v时观测到了0.2ma的电流(电流密度4ma·cm-2)(图20)。该时点的被加热体的电阻率是5.0×104ω·cm,这是表现出被加热体向等级b以上的电子晶体转变的情况。电压保持在75v的结果,约7秒之后观测到了约1100ma的电流的急剧增加,因而转换到上述电流控制(图21)。自成为上述电流控制后约100秒之后确认了试样的状态的结果,被加热体变成黑色且c12a7被电子晶体化了。上述被加热体中的电子浓度并没有差异,成为具有1.0×1021cm3的均匀的电子浓度的电子晶体。上述的被电子晶体化的被加热体(本实施例的电子晶体化钙铝石型化合物)的电传导率为5×102s/cm。
[实施例4]
以与实施例1相同的方式准备上述被加热体,当对上述试样施加电压时,将试样置于能够加热至最高1000℃的电加热炉中,并且将加热炉内部加热至1000℃。然后,使控制成最高10-29pa(10-34atom)为止的极低氧气分压的环境气体的ar(流量:3l/分钟、平均流速5.2cm/秒、大气压、室温)流动于加热炉内。利用极低氧气分压控制装置而从笼取出的氧混入于被极低氧气分压控制的ar环境气体中,而且利用氧气泵的原理从ar环境气体中提取该氧,并且使极低氧气分压状态下的ar重新流动于加热炉内。
在低温状态上述被加热体表现出绝缘体的行为,因此施加了最大100v的电压负载,但随着阶段性地升温产生了离子传导,并向试样流出电流而从0.1a·cm-2急速上升,因此将电压逐渐降低到10v。
在电压控制下,由于即使此后使温度上升也电流量不会增加,因此在温度上升到某种程度的阶段从电压控制转换到电流控制,据此流动于试样中的电流量与加热温度的上升一同增加,不久电阻就急剧下降,并下降到与金属传导体相同的水平。
测量此时的氩气中的氧浓度的话,则观察到了电阻急剧下降之后从试样中的显著的氧脱离。同时,在试样正极侧观察到了显著的发光现象。这意味着来自试样的脱离氧和连接在正极的钼线引起了氧化反应的情况。取出在电加热炉内自然冷却的试样的结果,确认到了原本透明的上述被加热体变成黑色且等级c的电子晶体化的情况、即,获得了电子浓度为1020/cm3以上的电子晶体。上述的被电子晶体化的被加热体(本实施例的电子晶体化钙铝石型化合物)的电传导率为5×102s/cm。
[比较例1]
将由fz法获得的c12a7单晶体切成10mm×5mm×1mm的大小并用作被加热体。利用如图3所示的装置而在加热炉中对该被加热体实施了加热处理。处理环境气体为极低氧气分压的ar环境气体(ar流量:3l/分钟、平均流速5.2cm/秒、大气压、常温)。
如图4所示,将该被加热体在1小时内从常温加热至900℃,然后在900℃下保持约25小时。之后,将上述被加热体升温至1000℃,进而保持5小时后,并使其自然冷却至常温。上述加热处理后的被加热体呈淡黄色。由此可知,上述加热处理后的钙铝石型化合物的电子浓度小于1.0×1017/cm3,其等级判定为a级。
由此发现仅通过加热处理不能实现钙铝石型化合物的充分的电子晶体化。
[比较例2]
除了代替比较例1的c12a7单晶而使用了多晶的c12a7:o2-粉末之外,按照如图5所示的履历进行了与比较例1相同的处理。与比较例1相同地,加热处理后的钙铝石型化合物的等级为a级。与c12a7单晶相同地,仅通过加热处理不能实现钙铝石型化合物的充分的电子晶体化。
(产业上的可利用性)
根据本发明的制备方法,与以往的方法相比可以缩短工序,进而以低温/短时间且较少能量来制备出高电子浓度的电子晶体。
- 还没有人留言评论。精彩留言会获得点赞!