一种球状二硫化钼及制备方法和其应用与流程
本发明属于无机纳米材料合成领域,具体涉及一种球状二硫化钼材料的制备方法,并将其应用于多环芳烃加氢过程中。
背景技术:
mos2具有良好的光、电、润滑、催化等性能,使其在电子探测、固体润滑、多相催化、储气材料及石油化工等方面有较好的应用潜力。mos2具有典型的层状结构,层与层之间以较弱的范德华力相结合,容易剥离,且层与层之间容易插入离子。单原子层中每个钼原子被六个硫原子所包围,呈兰角棱柱状,暴露出很多mo-s棱面,可作为催化活性中心。纳米尺寸的mos2具有量子尺寸效应、表面效应、暴露更多的活性中心等优点,使其具有更大的应用潜力。此外,纳米mos2的纯度、尺寸、形貌和晶相结构对其性能均具有较大的影响。因此,制备具有高纯相、可控形貌、尺寸的纳米mos2对开发高性能的mos2材料具有重要的意义。
球形纳米mos2具有较大的比表面积,较高的催化活性等优势成为人们研究的一个热点。目前,关于球形mos2的制备主要集中在溶液化学合成,尤其是水热或溶剂热方法。中国专利cn101851006a公布了一种以醇类做溶剂,采用多种钼源化合物(包括钼酸铵、钼酸纳、仲钼酸铵等)和硫源化合物(包括硫代乙酰胺、硫脲、硫、硫化铵等)做原料的超声辅助的溶剂热合成mos2的合成过程,可得到mos2微米球。中国专利cn103086436a公布了一种以二水合硫化钼,硫代乙酰胺为原料,无机盐k2cro4辅助的水热合成方法制备了由棒状结构的mos2堆积而成花状微米球。tian等以七钼酸铵,硫化纳作为原料,盐酸羟胺作为还原剂,十二烷基苯磺酸纳作表面活性剂,采用化学还原低温水热的合成过程制备了mos2纳米球(materialletter,2006,60,527-529)。在无机盐辅助和化学还原低温水热的过程中,产物中易残留杂原子,很难得到纯相的mos2。tang等以钼酸纳,硫脲为原料,以去离子水作为溶剂,ctab辅助的水热合成花状mos2微米球(micro&nanoletters,2013,164168)。mos2片层堆积的比较松散,球体表面粗糙,粒径不均一且颗粒间团聚严重。赵兴蕾等以钼酸钠和硫代乙酰胺为原料,以去离子水作为溶剂,在十六烷基溴化铵为表面活性剂,ph为8的条件下制得了mos2微米球。球体粒径不均一,团聚现象严重,且表面活性剂和ph对产物的结构、形貌影响较大。在强酸和碱性环境中,产物均为二氧化钼、三氧化钼和二硫化钼的混合物(青岛科技大学学报,2014,137-140.)水热或溶剂热过程中,硫源、钼源、溶剂、ph等条件对产物的结构,尺寸和形貌均有较大的影响。但水热合成的纳米级产物往往易团聚,尺寸和形貌难以调控。因此,水热合成高纯相的棒状纳米mos2具有非常重要的意义。
多环芳烃是一类含有两个或两个以上芳环的碳氢化合物,有些多环芳烃还含有氮、硫、氧等杂原子和环戊烷等结构单元。由于多环芳烃具有很强的致癌、致突变及致畸性,以及自身的疏水性及低水溶性能使其很快沉积到环境中,被公认为是威胁生态环境的主要污染物(wangwb.,etal.jacs,2003,125(35):10536-7.)。将多环芳烃加氢饱和后毒性会有所下降,且可以作为重要的化工原料和中间体广泛地应用于医药、染料以及燃料等领域。mos2作为催化剂的优点不仅仅是它的高活性,而且它对硫化物类毒物有很强的耐受性,不需要经过预硫化,催化剂稳定性好,不易失活。因此本发明制备的球状mos2在多环芳烃加氢领域具有广阔的应用前景。
技术实现要素:
本发明的目的是针对以上问题,提供了一种以多钼酸有机铵盐为钼源水热合成球状mos2的方法。多钼酸有机铵盐不溶于水,硫源中的硫逐步取代多钼酸有机铵盐中的氧形成中间体,随后该中间体进一步脱硫还原得到mos2。多钼酸有机铵盐前驱体既作为原料,也作为模板存在,硫源在多钼酸有机铵盐表面将其缓慢硫化还原原位生长mos2纳米片,使得mos2的生长得到了有效的控制。同时,这个过程中含硫生物试剂如l-半胱氨酸或谷胱甘肽均含有巯基,巯基具有还原性,可与钼源前驱体发生还原硫化反应,不需额外添加还原剂。由于棒状mos2表面由片层堆积而成,表面粗糙,暴露出大量的活性位点,在油品加氢催化方面有望得到良好的活性。该产物分离提纯也较为简单,产物mos2的收率可达理论收率的95%以上。
本发明提供的球状mos2的制备方法,步骤如下:
a.将钼盐溶于水中,滴加有机胺,调节溶液ph值在1~7之间,恒温静置数小时,自然冷却后得白色沉淀。将沉淀物过滤、洗涤、真空干燥,得到多钼酸有机铵盐前驱体;
b.配制悬浊液:溶剂中依次加入硫源、多钼酸有机铵盐,搅拌形成悬浊液。
c.溶剂热反应:将悬浊液转移至高压反应釜中,密封,置于烘箱中于120~200℃水热反应12~48小时。
d.分离洗涤:采用常规的分离手段,如抽滤,沉淀用去离子水和无水乙醇洗涤,真空干燥,得到黑色粉末状样品。
所述步骤a中钼盐选自七钼酸铵、钼酸纳中的一种或二者的混合物,钼盐在混合溶液中的浓度为0.05~0.25mol/l,优选0.05~0.15mol/l。
所述步骤a中有机胺为乙二胺或对苯二胺中的一种或二者的混合物,有机胺在混合溶液中的浓度为0.5~1.5mol/l,优选0.5~1mol/l。
所述步骤a中控制ph值在1~7之间,优选4~7,最优选4~5。
所述步骤a中恒温温度为50~90℃,优选50~70℃,静置反应时间0.5~4小时,优选0.5~1.5小时。
所述步骤b中硫源为l-半胱氨酸或谷胱甘肽或二者的混合物,硫源浓度为0.02~0.5mol/l,优选0.1~0.3mol/l,原料中s/mo摩尔比为10:1~2:1。
所述步骤b中悬浊液的ph范围为1~7之间,优选4~6。
所述步骤b中溶剂为水或乙醇或n,n-二甲基甲酰胺或任意两种的混合液或三种的混合液。
所述步骤c中溶剂热反应的温度为120~240℃,优选160~200℃,反应时间为12~48小时,优选16~24小时。
所述真空干燥条件:温度40℃~70℃,优选60℃~70℃,时间:6~14小时,优选8~12小时。
附图说明
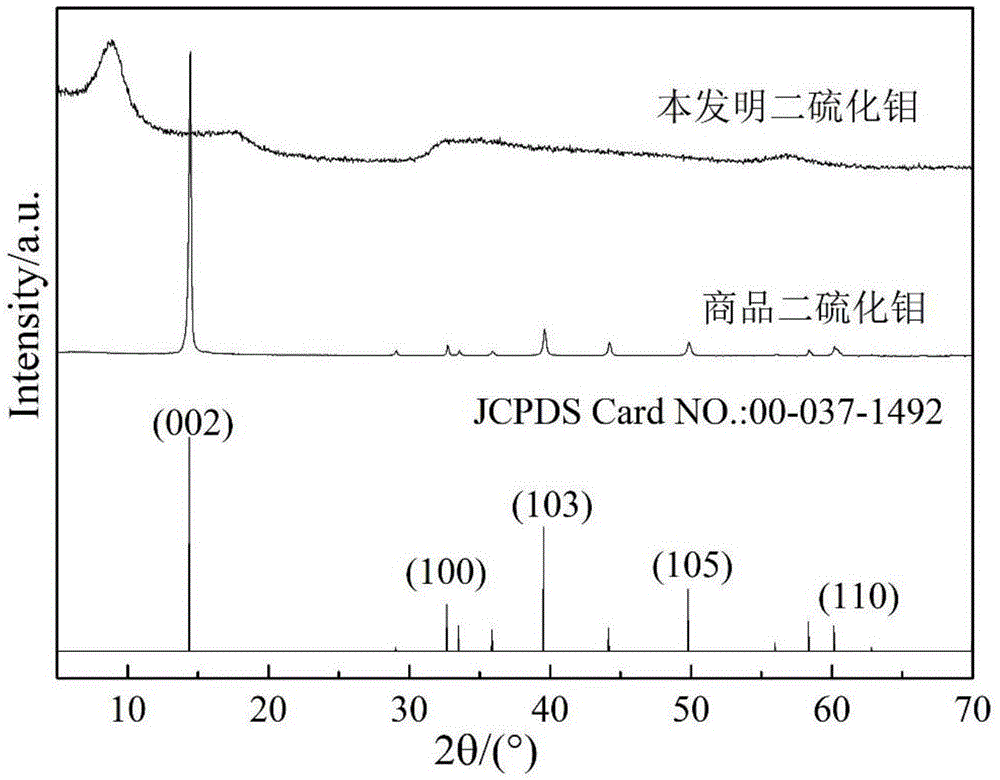
图1是实施例1中得到的球状mos2的xrd图;
图2a是实施例1中得到的球状mos2的低倍tem图;
图2b是实施例1中得到的球状mos2的高倍tem图;
具体实施方式
下面结合具体实施例对本发明做进一步的具体说明,但本发明不受下述实施例的限制。任何不超出本发明的构思和范畴的改动,均在本发明范围内。
实施例1:
一种球状mos2纳米材料的制备方法,步骤包括:
步骤一:称取1mmol钼酸铵溶解于15ml去离子水中,向溶液中滴加14mmol乙二胺,300rpm搅拌速率下滴加1mol/l盐酸溶液至溶液ph为4~5,得白色沉淀,于50℃恒温静置1小时,沉淀用无水乙醇和去离子水洗涤,然后在得70℃下真空干燥12小时得前驱体三钼酸乙二胺[(c2h10n2)mo3o10]。
步骤二:称取8mmoll-半胱氨酸溶解于25ml去离子水和n,n-二甲基甲酰胺1:1的混合溶液中,形成0.32mol/l的溶液。再加入0.7mmol上述三钼酸乙二胺,调节溶液ph为5~6。充分搅拌后将悬浊液转移至100ml水热反应釜中,于200℃水热反应16小时,自然冷却后,抽滤,沉淀用去离子水和无水乙醇洗涤,然后在70℃下真空干燥12小时,得到黑色粉末样品。制备得到的黑色粉末分别进行xrd(x射线粉末衍射)、sem(扫描电子显微镜)和tem(透射电子显微镜)表征。xrd谱图表明产物是mos2,衍射峰峰宽变大,表明所得mos2尺寸较小。tem图像表明产物为直径500~1000nmmos2微米球(参见图2a、图2b),微米球表面由大量mos2纳米片堆积而成。高倍tem图像表明产物是由大量弯曲的mos2片层(线条状结构)相互交联堆积而成,每个片层的长度在几十个纳米左右(参见图2b)。
实施例2:
一种球状mos2纳米材料的制备方法,步骤包括:
步骤一:称取1mmol钼酸铵溶解于15ml去离子水中,向溶液中滴加14mmol对苯二胺,300rpm搅拌速率下滴加1mol/l盐酸溶液至溶液ph为4~5,得白色沉淀,于50℃恒温静置1小时得三钼酸对苯二胺((c6h10n2)mo3o10)。
步骤二:称取8mmol谷胱甘肽溶解于50ml去离子水和乙醇1:1的混合溶液中,形成0.16mol/l的溶液,再加入0.7mmol上述三钼酸对苯二胺,调节溶液ph为5~6。充分搅拌后将悬浊液转移至高压反应釜中,于200℃水热反应16小时,自然冷却后,抽滤,沉淀用去离子水和无水乙醇洗涤,然后在70℃下真空干燥12小时,得到黑色粉末样品。xrd谱图表明产物是mos2,衍射峰峰宽变大,表明所得mos2尺寸较小。tem图像表明产物为直径500~1000nmmos2微米球,微米球表面由大量mos2纳米片堆积而成。
实施例3:
一种球状mos2纳米材料的制备方法,步骤包括:
步骤一:称取1mmol钼酸铵溶解于15ml去离子水中,向溶液中滴加14mmol乙二胺,300rpm搅拌速率下滴加1mol/l盐酸溶液至溶液ph为1~2,得白色沉淀,于50℃陈化1小时,沉淀用无水乙醇和去离子水洗涤,然后在得70℃下真空干燥12小时得三钼酸乙二胺[(c2h10n2)mo3o10]。
步骤二:称取8mmoll-半胱氨酸溶解于50ml去离子水中,形成0.16mol/l的溶液,再加入0.7mmol上述三钼酸乙二胺,调节溶液ph为5~6。充分搅拌后将悬浊液转移至高压反应釜中,于200℃水热反应16小时,自然冷却后,抽滤,沉淀用去离子水和无水乙醇洗涤,然后在70℃下真空干燥12小时,得到黑色粉末样品。xrd谱图表明产物是mos2,衍射峰峰宽变大,表明所得mos2尺寸较小。tem图像表明产物为直径500~1000nmmos2微米球,微米球表面由大量mos2纳米片堆积而成。
实施例4:
一种球状mos2纳米材料的制备方法,步骤包括:
步骤一:称取1mmol钼酸钠溶解于15ml去离子水中,向溶液中滴加14mmol乙二胺,300rpm搅拌速率下滴加1mol/l盐酸溶液至溶液ph为3~4,得白色沉淀,于50℃恒温静置1小时,沉淀用无水乙醇和去离子水洗涤,然后在得60℃下真空干燥12小时得前驱体三钼酸乙二胺[(c2h10n2)mo3o10]。
步骤二:称取8mmoll-半胱氨酸溶解于25ml去离子水中,形成0.32mol/l的溶液,再加入0.7mmol上述三钼酸乙二胺,调节溶液ph为5~6。充分搅拌后将悬浊液转移至高压反应釜中,于200℃水热反应16小时,自然冷却后,抽滤,沉淀用去离子水和无水乙醇洗涤,然后在70℃下真空干燥12小时,得到黑色粉末样品。xrd谱图表明产物是mos2,衍射峰峰宽变大,表明所得mos2尺寸较小。tem图像表明产物为直径500~1000nmmos2微米球,微米球表面由大量mos2纳米片堆积而成。
实施例5:
一种球状mos2纳米材料的制备方法,步骤包括:
步骤一:称取1mmol钼酸钠溶解于15ml去离子水中,向溶液中滴加7mmol乙二胺,300rpm搅拌速率下滴加2mol/l盐酸溶液至溶液ph为3~4,得白色沉淀,于50℃恒温静置1小时,沉淀用无水乙醇和去离子水洗涤,然后在得60℃下真空干燥12小时得前驱体三钼酸乙二胺[(c2h10n2)mo3o10]。
步骤二:称取8mmoll-半胱氨酸溶解于25ml去离子水中,形成0.32mol/l的溶液,再加入0.7mmol上述三钼酸乙二胺,调节溶液ph为5~6。充分搅拌后将悬浊液转移至高压反应釜中,于200℃水热反应16小时,自然冷却后,抽滤,沉淀用去离子水和无水乙醇洗涤,然后在70℃下真空干燥12小时,得到黑色粉末样品。xrd谱图表明产物是mos2,衍射峰峰宽变大,表明所得mos2尺寸较小。tem图像表明产物为直径500~1000nmmos2微米球,微米球表面由大量mos2纳米片堆积而成。
实施例6:
一种球状mos2纳米材料的制备方法,步骤包括:
实施例6与实施例1的制备方法相同,不同之处在于步骤二中的水热反应时间为48小时。得到黑色粉末样品,xrd谱图表明产物是mos2,衍射峰峰宽变大,表明所得mos2尺寸较小。tem图像表明产物为直径500~1000nmmos2微米球(参见图2a、图2b),微米球表面由大量mos2纳米片堆积而成。
实施例7:
一种球状mos2纳米材料的制备方法,步骤包括:
实施例7与实施例1的制备方法相同,不同之处在于步骤二中的水热反应温度为160℃。得到黑色粉末样品,xrd谱图表明产物是mos2,衍射峰峰宽变大,表明所得mos2尺寸较小。tem图像表明产物为直径500~1000nmmos2微米球,微米球表面由大量mos2纳米片堆积而成。
实施例8:
一种球状mos2纳米材料的制备方法,步骤包括:
实施例8与实施例1的制备方法相同,不同之处在于步骤二中调节溶液ph为1.5。得到黑色粉末样品,xrd谱图表明产物是mos2,衍射峰峰宽变大,表明所得mos2尺寸较小。tem图像表明产物为直径500~1000nmmos2微米球,微米球表面由大量mos2纳米片堆积而成。
实施例9:
将实施例1-8制备得到的产物用做催化剂,对其进行蒽加氢催化性能的评估,并用商用mos2做对比,步骤如下:在悬浮床反应体系的l00ml高压釜反应器中加入0.075g实施例1制备的mos2催化剂(基于蒽重量百分比为2.5%),再加入蒽3g和正十三烷30g。装好高压釜后用氢气置换空气3次(每次先关闭尾气阀,然后打开进气阀,以l00ml/min的氢气升压至2mpa后关闭进气阀,再打开尾气阀排空。),升压到8mpa,打开搅拌,搅拌速度为300r/min。10℃/min升温至350℃时开始计时,保持4小时后自然降温。
蒽催化加氢评价结果如表1。
表1mos2催化剂的蒽催化加氢评价结果
下面结合表1和实施例1对本发明做进一步详细说明。蒽催化加氢反应结果包括产物选择性、蒽转化率和蒽的加氢率,加氢产物分别为二氢蒽(h2a)、四氢蒽(h4a)、八氢蒽(h8a)、十四氢蒽(h14a)。
本发明的mos2催化剂用于蒽悬浮床加氢反应中,蒽的转化率为99.8%,深度加氢产物八氢蒽选择性最高达87%,是商品mos2催化剂的7.3倍:加氢率最高达57%,是商品mos2的2.4倍(参见表1)。
- 还没有人留言评论。精彩留言会获得点赞!