一种钙钛矿CsPbBr3量子点及其制备方法和应用与流程
本发明涉及发光材料技术领域,更具体地,涉及一种钙钛矿cspbbr3量子点及其制备方法和应用。
背景技术:
铅卤钙钛矿量子点因其极高的发光色纯度、发光效率、较高的载流子迁移率、高的量子效率等特点,在光电探测、太阳能电池、照明、显示等领域有着广泛的应用。
目前制备钙钛矿量子点的方法主要有热注入法与过饱和重结晶法。热注入法需要苛刻的合成环境,制备过程中需要高温环境下,且还需要通入惰性气体进行保护,制备难度大,合成效率低,也不利于规模化生产。过饱和重结晶法是通过将钙钛矿前驱体溶液加入不良溶剂中,在表面活性剂的辅助下形成钙钛矿量子点。然而,不同卤素配比的钙钛矿量子点混合之后容易发生阴离子交换,使得各自的成分与发光波长发生改变,这限制了其在显示领域的应用。
因此,需要开发出制备钛矿量子点过程操作步骤简单、条件温和且产物稳定的方法。
技术实现要素:
本发明为克服上述现有技术所述的需要高温环境、需要惰性气体保护、制备过程复杂、难度大、难以规模化生产和产物稳定性不足的缺陷,提供一种钙钛矿cspbbr3量子点的制备方法,所述制备方法操作步骤简单,条件温和,不需要保护气体,重复率高,可规模化生产,产物稳定,具有高效荧光的特点。
本发明的另一目的在于,提供上述制备方法制得的钙钛矿cspbbr3量子点。
本发明的还一目的在于,提供上述钙钛矿cspbbr3量子点在制备光电器件中的应用。
为解决上述技术问题,本发明采用的技术方案是:
一种钙钛矿cspbbr3量子点的制备方法,包括如下特征:
s1.制备含有csbr、pbbr2、二甲基甲酰胺、油酸和油胺的混合溶液;
s2.在步骤s1.的混合溶液中,加入硅球颗粒,然后30~60℃搅拌反应至少1h,得到所述钙钛矿cspbbr3量子点。
发明人偶然发现,硅球颗粒能够作为固体催化剂,快速制备钙钛矿cspbbr3量子点。钙钛矿量子点作为一种离子晶体,由于反应动力学快,其具有独特的生长特性。加入大颗粒硅球,使得成核动力学速度更快。较高的反应温度尽管会加快反应速度,也会显著削弱配体(如油酸,油胺)与材料表面的结合,降低材料的键合效果。过低的反应温度不利于材料的成核过程。
上述制备方法在制备过程中仅仅需要添加一种固体催化剂硅球颗粒,能够快速沉淀出钙钛矿cspbbr3量子点,操作步骤简单,反应时间短,条件温和,不需要保护气体,重复率高,可规模化生产;而且,产物稳定,钙钛矿原有的结构不会被破坏,所得的产物仍能维持原有的高效荧光的特点。该制备方法避免了传统保护钙钛矿量子点方法中所需的较高反应温度、长时间反应、破坏表面等不足。
优选地,所述硅球颗粒的质量与混合溶液的体积之比为0.008~0.024g/ml。而且,发明人研究发现,增加硅球颗粒的加入量时,制得的钙钛矿cspbbr3量子点的发射峰向短波处移动。
优选地,所述硅球颗粒的质量与混合溶液的体积之比为0.016g/ml。
优选地,步骤s2.中搅拌反应的温度为60℃,时间为2h。
优选地,所述硅球颗粒的粒径为150~300nm。
所述硅球颗粒可由本领域技术人员参考现有技术制备得到。用已知的stober法制备得到。具体地,所述硅球颗粒可通过如下方法制备得到:取23.5ml超纯水,63.5ml醇溶液,13ml氨水混合,并逐滴滴加0.6ml正硅酸乙酯,在35℃下剧烈搅拌30min形成硅球种子溶液。然后逐滴滴加5ml正硅酸乙酯,在35℃剧烈搅拌2h得乳白色浑浊溶液。用无水乙醇和超纯水离心洗涤后,放到90℃干燥箱中干燥24h,得到硅球颗粒。所述醇溶液可以为甲醇、乙醇、异丙醇或正丁醇中的一种或两种以上的组合的水溶液。
优选地,所述csbr与pbbr2的摩尔比为(0.8~1.2)∶1。更优选地,所述csbr与pbbr2的摩尔比为1∶1。
优选地,所述混合溶液中csbr的摩尔浓度为0.032~0.048mmol/ml。更优选地,所述混合溶液中csbr的摩尔浓度为0.04mmol/ml。
优选地,所述混合溶液中pbbr2的摩尔浓度为0.032~0.048mmol/ml。更优选地,所述混合溶液中pbbr2的摩尔浓度为0.04mmol/ml。
优选地,所述混合溶液中二甲基甲酰胺、油酸和油胺的体积比为1∶(0.1~0.2)∶(0.04~0.1)。更优选地,所述混合溶液中二甲基甲酰胺、油酸和油胺的体积比为1∶0.18∶0.06。
优选地,所述搅拌反应的搅拌速率为800~1000r/min。
优选地,所述制备方法还包括后处理的步骤,所述后处理为:将搅拌反应后得到的沉淀进行离心洗涤、烘干。所述离心洗涤可以采用乙酸乙酯进行离心洗涤。所述离心洗涤的转速为8000~10000r/min,时间为4~6min。所述烘干的条件可以为在40℃干燥箱中干燥24h。
本发明同时保护上述制备方法制得的钙钛矿cspbbr3量子点。
本发明还保护上述钙钛矿cspbbr3量子点在制备光电器件中的应用。
与现有技术相比,本发明的有益效果是:
本发明提供的制备方法在制备过程中仅仅需要添加一种固体催化剂硅球颗粒,能够快速沉淀出钙钛矿cspbbr3量子点,操作步骤简单,反应时间短,条件温和,不需要保护气体,重复率高,可规模化生产;而且,产物稳定,钙钛矿原有的结构不会被破坏,所得的产物仍能维持原有的高效荧光的特点。
附图说明
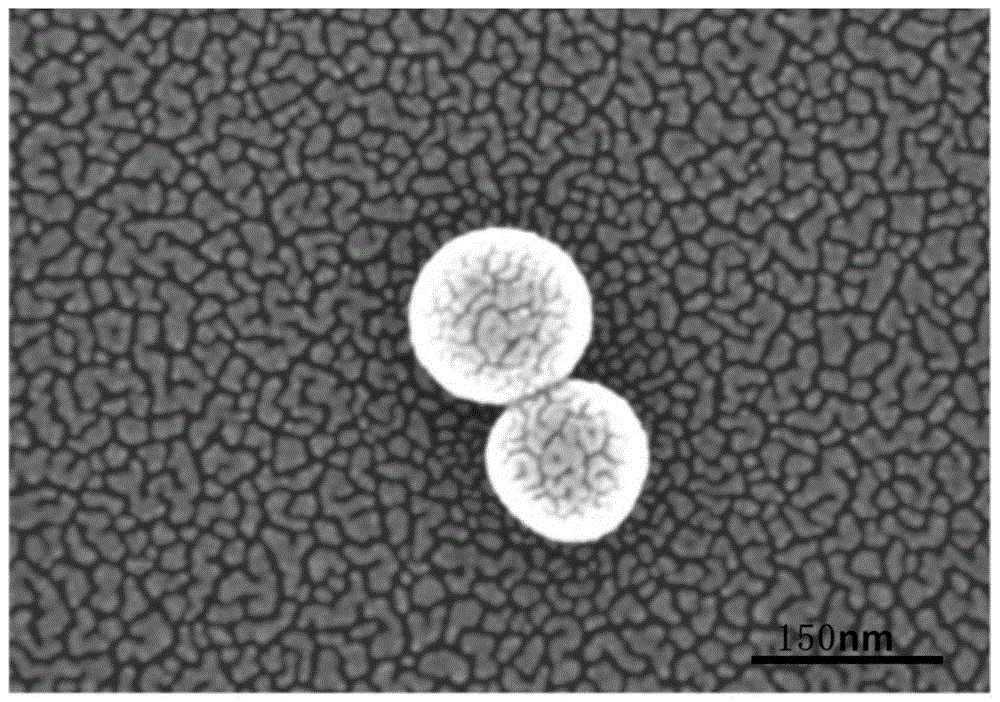
图1为实施例2制得的钙钛矿cspbbr3量子点的sem扫描电镜图。
图2为实施例1~3制得的钙钛矿cspbbr3量子点在激发波长为365nm下的荧光发射光谱。
图3为实施例1制得的钙钛矿cspbbr3量子点在监测波长为514nm下的荧光激发光谱。
图4为实施例3制得的钙钛矿cspbbr3量子点粉末的宏观照片。
图5为实施例3制得的钙钛矿cspbbr3量子点粉末在265nm紫外光下的发光照片。
具体实施方式
下面结合具体实施方式对本发明作进一步的说明。
实施例中的原料均可通过市售得到;
除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
实施例1~3中,硅球颗粒通过如下方法制备得到:取23.5ml超纯水,63.5ml甲醇溶液,13ml氨水混合,并逐滴滴加0.6ml正硅酸乙酯,在35℃下剧烈搅拌30min。然后逐滴滴加5ml正硅酸乙酯,在35℃剧烈搅拌2h得乳白色浑浊溶液。用无水乙醇和超纯水以转速9000r/min离心洗涤5min后,放到90℃干燥箱中干燥24h,收集得到硅球颗粒。制得的硅球颗粒的粒径为150~300nm。
实施例1
本实施例添加0.1g固体催化剂硅球颗粒。
一种钙钛矿cspbbr3量子点的制备方法,步骤如下:
s1.将0.147g金属卤化盐pbbr2和0.085gcsbr称量好放入50ml菌种瓶中,然后加入10mldmf,再加入1.8ml油酸和0.6ml油胺形成混合溶液。
s2.取0.1g硅球颗粒加入步骤s1.的混合溶液中,在60℃水浴下以转速800~1000r/min搅拌2h得沉淀物,用乙酸乙酯以转速9000r/min离心洗涤5min收集沉淀物,即得钙钛矿cspbbr3量子点。在40℃干燥箱中干燥24h,即得本发明中用简易方法合成的钙钛矿cspbbr3量子点。
实施例2
本实施例添加0.2g固体催化剂硅球颗粒。
本实施例制备钙钛矿cspbbr3量子点的步骤如下:
s1.将0.147g金属卤化盐pbbr2和0.085gcsbr称量好放入50ml菌种瓶中,然后加入10mldmf,再加入1.8ml油酸和0.6ml油胺形成混合溶液。
s2.取0.2g硅球颗粒加入步骤s1.的混合溶液中,在60℃水浴下以转速800~1000r/min搅拌2h得沉淀物,用乙酸乙酯以转速9000r/min离心洗涤5min收集沉淀物,即得钙钛矿cspbbr3量子点。在40℃干燥箱中干燥24h,即得本发明中用简易方法合成的钙钛矿cspbbr3量子点。
实施例3
本实施例添加0.3g固体催化剂硅球颗粒。
本实施例制备钙钛矿cspbbr3量子点的步骤如下:
s1.将0.147g金属卤化盐pbbr2和0.085gcsbr称量好放入50ml菌种瓶中,然后加入10mldmf,再加入1.8ml油酸和0.6ml油胺形成混合溶液。
s2.取0.3g硅球颗粒加入步骤s1.的混合溶液中,在60℃水浴下以转速800~1000r/min搅拌2h得沉淀物,用乙酸乙酯以转速9000r/min离心洗涤5min收集沉淀物,即得钙钛矿cspbbr3量子点。在40℃干燥箱中干燥24h,即得本发明中用简易方法合成的钙钛矿cspbbr3量子点。
测试与表征
(1)sem扫描电镜分析
扫描电镜形貌表征(sem)采用日本hitachsu8220场发射扫描电子显微镜。
(2)荧光激发光谱和荧光发射光谱
激发光谱检测采用爱丁堡fls-980荧光光谱仪;发射光谱检测采用爱丁堡fls-980荧光光谱仪。
测试结果
图1是实施例2制得的钙钛矿cspbbr3量子点的sem扫描电镜图,证实了所得钙钛矿量子点是纳米材料,粒径为100~150nm。
在激发波长为365nm下,实施例1制得的钙钛矿cspbbr3量子点的发射光谱如图2所示,发射峰在514nm。在监测波长为514nm下,研究实施例1制得的钙钛矿cspbbr3量子点的激发光谱,如图3所示。
在激发波长为365nm下,实施例2制得的钙钛矿cspbbr3量子点的发射光谱如图2所示,发射峰在494nm。
在激发波长为365nm下,实施例3制得的钙钛矿cspbbr3量子点的发射光谱如图2所示,发射峰在475nm。图4是实施例3制得的钙钛矿cspbbr3量子点的照片,图5是实施例3制得的钙钛矿cspbbr3量子点在254nm紫外光照射下的发光照片。
根据图2可知,添加的硅球颗粒越多,钙钛矿cspbbr3量子点的发射峰向短波处移动。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!