由蚕豆制备碳点的方法、碳点及其应用与流程
本发明涉及碳纳米材料领域,尤其涉及一种由蚕豆制备碳点的方法、碳点及其应用。
背景技术:
碳点是一种新型的纳米材料,近年来受到广大科研工作者的广泛关注。碳点是指尺寸小于10nm的碳球状纳米粒子,它具有特征性的荧光激发依赖性。与传统的半导体量子点相比,它具有水溶性好、化学稳定性高、易功能化、耐光漂白、低毒、生物相容性好等优点。这些优点使得碳点可应用于生物医学领域包括生物成像、生物传感器、生物医学交付系统等,还可应用于光电子学领域包括染料敏华的太阳能电池、有机太阳能电池、超级电容器、发光器件等。
现有技术中用于制备碳点的碳源包括液体和固体两类,其中液体有橘汁、草莓汁、苹果汁、香蕉汁、豆浆和牛奶等;固体有草、菌类、花粉、壳聚糖、土豆、冬瓜、明胶、大蒜、木瓜、胖大海种子、姜和萝卜等。
迄今为止,尚未见以蚕豆为碳源制备碳点的方法报道。
技术实现要素:
有鉴于此,本发明的目的在于提供一种由蚕豆制备碳点的方法、碳点及其应用,合成方法简单有效,原料廉价易得,反应条件温和可控,且环境友好,在一般的实验室均能完成,易于推广,且制备的碳点荧光量子产率较高,且具有良好的荧光性能。
为实现以上目的,本发明提供技术方案如下:
一种由蚕豆制备碳点的方法,包括:
提供蚕豆粉;
将蚕豆粉和水混合得到混合物进行水热反应;
将经所述水热反应得到的产物进行固液分离,及对得到的液体进行透析,得到碳点溶液。
作为上述技术方案的进一步改进,所述蚕豆粉和水的用量比为:(6~10)g:(30~50)ml。
作为上述技术方案的进一步改进,所述方法还包括:在进行所述水热反应前,向所述混合物中加入氮源;优选地,所述氮源包括尿素、二乙醇胺、乙二胺和三聚氰胺中的一种或几种。
作为上述技术方案的进一步改进,所述蚕豆粉和氮源的质量比为:(3~6):(1.2~2.4)。
作为上述技术方案的进一步改进,所述蚕豆粉通过将蚕豆研磨成粉得到;所述方法还包括:在对所述蚕豆进行研磨前,对所述蚕豆进行清洗。
作为上述技术方案的进一步改进,所述方法还包括:在进行所述水热反应前,将所述混合物进行超声处理。
作为上述技术方案的进一步改进,所述水热反应的温度为180~200℃,所述水热反应的时间为10~14h。
作为上述技术方案的进一步改进,所述固液分离包括:将经所述水热反应得到的产物冷却至室温后进行离心,得到的上清液;优选地,所述离心转速为10000~12000r/min,所述离心时间为10~20min。
作为上述技术方案的进一步改进,所述透析采用的透析袋的截留分子量为500~1000;优选地,所述透析时间为20~24h;更优地,在所述透析过程中同时对所述透析袋进行搅拌。
作为上述技术方案的进一步改进,所述方法还包括:对所述碳点溶液进行冷冻干燥。
本发明还提供了一种上述的由蚕豆制备碳点的方法制备得到的碳点。
本发明还提供了一种上述的碳点在对水中hg2+的检测中的应用。
本发明还提供了一种上述的碳点在细胞成像中的应用。
本发明的有益效果:
本发明以廉价来源易得的生物质蚕豆为碳源,通过采用一步水热法合成碳点,该合成方法简单有效,原料廉价易得,反应条件温和可控,且环境友好,在一般的实验室均能完成,易于推广。采用本发明方法制备的碳点的荧光量子产率较高,具有良好的荧光性能,可用于对水中的hg2+进行检测;该碳点还具有光稳定性好、毒性低、生物相容性好等优点,在细胞成像方面也具有良好的应用前景。
进一步地,通过向碳源中加入氮源进行氮掺杂,制备得到的氮掺杂碳点较未掺杂的碳点的荧光量子产率明显更高,可达12%以上,且具有更好的荧光性能;能够更好地应用于对水中的hg2+进行检测,及细胞成像。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对本发明范围的限定。
图1为本发明实施例1制备的cds的tem图;
图2为本发明实施例4制备的n-cds的tem图;
图3为本发明实施例1制备的cds的紫外可见吸收光谱图;
图4为本发明实施例4制备的n-cds的紫外可见吸收光谱图;
图5为本发明实施例1制备的cds的荧光光谱图;
图6为本发明实施例4制备的n-cds的荧光光谱图;
图7为本发明实施例4制备的n-cds在不同波长光激发下的荧光发射光谱图;
图8为本发明实施例1制备的cds的红外光谱图;
图9为本发明实施例4制备的n-cds的红外光谱图;
图10为本发明实施例1制备的cds的xrd图;
图11为本发明实施例4制备的n-cds的xrd图;
图12为本发明实施例1制备的cds的xps图;
图13为本发明实施例4制备的n-cds的xps图;
图14为不同浓度的hg2+滴定本发明实施例4制备的n-cds的荧光猝灭曲线图;
图15为不同浓度hg2+滴定本发明实施例4制备的n-cds的强度的峰值的拟合工作曲线;
图16为本发明实施例4制备的n-cds对不同金属离子的选择性竞争图;
图17为本发明实施例4制备的n-cds的荧光强度与ph关系图;
图18为本发明实施例4制备的n-cds对细胞毒性实验结果图;
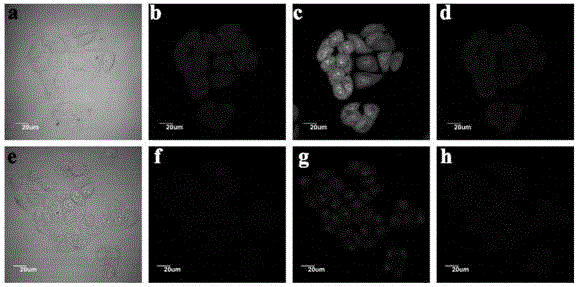
图19为本发明实施例4制备的n-cds在hela细胞中的荧光成像图及n-cds在hela细胞中对hg2+的响应图:(a)hela细胞的亮场图像,(b)n-cds标记hela细胞的荧光成像(激发波长为405nm),(c)n-cds标记hela细胞的荧光成像(激发波长为488nm),(c)n-cds标记hela细胞的荧光成像(激发波长为559nm),(e)hela细胞加入hg2+的亮场图像,(f)加入hg2+的n-cds标记hela细胞的荧光成像(激发波长为405nm),(g)加入hg2+的n-cds标记hela细胞的荧光成像(激发波长为488nm),(h)加入hg2+的n-cds标记hela细胞的荧光成像(激发波长为559nm)。
具体实施方式
如本文所用之术语:
“由……制备”与“包含”同义。本文中所用的术语“包含”、“包括”、“具有”、“含有”或其任何其它变形,意在覆盖非排它性的包括。例如,包含所列要素的组合物、步骤、方法、制品或装置不必仅限于那些要素,而是可以包括未明确列出的其它要素或此种组合物、步骤、方法、制品或装置所固有的要素。
连接词“由……组成”排除任何未指出的要素、步骤或组分。如果用于权利要求中,此短语将使权利要求为封闭式,使其不包含除那些描述的材料以外的材料,但与其相关的常规杂质除外。当短语“由……组成”出现在权利要求主体的子句中而不是紧接在主题之后时,其仅限定在该子句中描述的要素;其它要素并不被排除在作为整体的所述权利要求之外。
当量、浓度、或者其它值或参数以范围、优选范围、或一系列上限优选值和下限优选值限定的范围表示时,这应当被理解为具体公开了由任何范围上限或优选值与任何范围下限或优选值的任一配对所形成的所有范围,而不论该范围是否单独公开了。例如,当公开了范围“1~5”时,所描述的范围应被解释为包括范围“1~4”、“1~3”、“1~2”、“1~2和4~5”、“1~3和5”等。当数值范围在本文中被描述时,除非另外说明,否则该范围意图包括其端值和在该范围内的所有整数和分数。
在这些实施例中,除非另有指明,所述的份和百分比均按质量计。
“质量份”指表示多个组分的质量比例关系的基本计量单位,1份可表示任意的单位质量,如可以表示为1g,也可表示2.689g等。假如我们说a组分的质量份为a份,b组分的质量份为b份,则表示a组分的质量和b组分的质量之比a:b。或者,表示a组分的质量为ak,b组分的质量为bk(k为任意数,表示倍数因子)。不可误解的是,与质量份数不同的是,所有组分的质量份之和并不受限于100份之限制。
“和/或”用于表示所说明的情况的一者或两者均可能发生,例如,a和/或b包括(a和b)和(a或b)。
本发明提供一种由蚕豆制备碳点的方法,包括:
提供蚕豆粉;
将蚕豆粉和水混合得到混合物进行水热反应;
将经所述水热反应得到的产物进行固液分离,然后对经所述固液分离得到的液体进行透析,得到碳点溶液。
需要说明的是,上述水热反应在具有聚四氟乙烯内衬的不锈钢水热反应釜中进行。上述水可采用去离子水、超纯水或蒸馏水。
进一步地,所述蚕豆粉和水的用量比为:(6~10)g:(30~50)ml。
优选地,所述方法还包括:在进行所述水热反应前,向所述混合物中加入氮源。
本发明进一步通过将向蚕豆粉和水的混合物氮源,然后通过水热反应等可制备得到氮掺杂碳点(n-cds),其粒径相对无掺杂的碳点(cds)的粒径更小,具有更高的量子产率及更好的荧光效果。
可选地,所述氮源为尿素、二乙醇胺、乙二胺和三聚氰胺中的一种或几种,即氮源可以为尿素、二乙醇胺、乙二胺和三聚氰胺中的任意一种,氮源也可以为尿素、二乙醇胺、乙二胺和三聚氰胺中的至少两种按任意比例的混合物。
进一步地,所述蚕豆粉和氮源的质量比为:(3~6):(1.2~2.4)。
优选地,所述蚕豆粉通过将蚕豆研磨成粉得到;所述方法还包括:在对所述蚕豆进行研磨前,对所述蚕豆进行清洗及干燥。需要说明的是,在研磨前先对蚕豆进行清洗优选采用超声清洗,可将蚕豆的褶皱里的灰尘清洗干净。上述干燥可采用自然晾干或低温热风烘干等方式。
优选地,所述方法还包括:在进行所述水热反应前,将所述混合物置于超声机中进行超声。上述超声的目的是除去混合物中的气泡;进一步地,还可在将上述混合物置于超声机边进行超声的同时边用玻璃棒进行搅拌,使蚕豆粉在水中分散更均匀,以防止结块影响后续的水热反应。
可选地,所述水热反应的温度为180~200℃,所述水热反应的时间为10~14h。
进一步地,所述固液分离包括:将经所述水热反应得到的产物冷却至室温后进行离心,然后将经所述离心得到的上清液收集于透析袋中。优选地,所述离心转速为10000~12000r/min,所述离心时间为10~20min。当离心转速为10000~12000r/min,离心时间为10~20min时,固液分离效果更好。
进一步地,所述透析采用的透析液为去离子水、纯水或超纯水;采用的透析袋的截留分子量为500~1000,采用截留分子量为500~1000的透析袋才能对离心得到的第二上清液进行较好地透析纯化,去除杂质。所述透析过程在室温条件下进行。可选地,所述透析时间为20~24h。优选地,在所述透析过程中同时对所述透析袋进行采用400~60r/min的速度缓慢搅拌,可以加速透析效果;且每间隔6~8h更换一次透析液。
进一步地,所述方法还包括:对所述碳点溶液进行冷冻干燥,得到褐色的碳点粉末(cds)。
本发明还提供了一种上述的由蚕豆制备碳点的方法制备得到的碳点。
本发明以廉价来源易得的生物质蚕豆为碳源,通过采用一步水热法合成碳点,该合成方法简单有效,原料廉价易得,反应条件温和可控,且环境友好,在一般的实验室均能完成,易于推广。采用本发明方法制备的碳点的荧光量子产率可达4%以上,具有良好的荧光性能,可用于对水中的hg2+进行检测;该碳点还具有光稳定性好、毒性低、生物相容性好等优点,在细胞成像方面也具有良好的应用前景。
进一步地,通过向碳源中加入氮源进行氮掺杂,制备得到的氮掺杂碳点较未掺杂的碳点的荧光量子产率明显更高,可达12%以上,且具有更好的荧光性能;能够更好地应用于对水中的hg2+进行检测,且检测范围较宽及检测限较低;且能够更好地应用于细胞成像。
下面将结合具体实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
实施例1
(1)取青海省天然蚕豆,先采用超声清洗干净,然后自然晾干并研磨成粉末,得到蚕豆粉;
(2)取6g蚕豆粉置于聚四氟乙烯内衬中,然后添加30ml去离子水,再将聚四氟乙烯内衬置于超声清洗器中边超声,边用玻璃棒将蚕豆粉与去离子水搅拌混合均匀得到分散均匀的混合物。
(3)将上述聚四氟乙烯内衬密封于不锈钢反应釜中,拧紧不锈钢反应釜盖,放入200℃恒温干燥箱中反应10h后将反应釜取出,放于通风橱中使反应产物自然冷却至室温。
(4)将上述冷却后的反应产物置于离心管中,在10000r/min转速下离心20min;然后用移液枪将离心得到的上清液转移至截留分子量为1000的透析袋中,并将透析袋置于去离子水中在室温条件下进行透析,同时用60r/min的速度缓慢搅拌透析袋,每间隔8h更换一次去离子水,共透析24h,最后收集透析袋内的液体得到碳点溶液。
(5)将碳点溶液进行冷冻干燥得到的褐色粉末,即为碳点(cds)。
实施例2
(1)取青海省天然蚕豆,先采用超声清洗干净,然后自然晾干并研磨成粉末,得到蚕豆粉;
(2)取8g蚕豆粉置于聚四氟乙烯内衬中,然后添加40ml去离子水,再将聚四氟乙烯内衬置于超声清洗器中边超声,边用玻璃棒将蚕豆粉与去离子水搅拌混合均匀得到分散均匀的混合物。
(3)将上述聚四氟乙烯内衬密封于不锈钢反应釜中,拧紧不锈钢反应釜盖,放入180℃恒温干燥箱中反应14h后将反应釜取出,放于通风橱中使反应产物自然冷却至室温。
(4)将上述冷却后的反应产物置于离心管中,在12000r/min转速下离心10min;然后用移液枪将离心得到的上清液转移至截留分子量为800的透析袋中,并将透析袋置于去离子水中在室温条件下进行透析,同时用50r/min的速度缓慢搅拌透析袋,每间隔7h更换一次去离子水,共透析21h,最后收集透析袋内的液体得到碳点溶液。
(5)将碳点溶液进行冷冻干燥得到的褐色粉末,即为碳点(cds)。
实施例3
(1)取青海省天然蚕豆,先采用超声清洗干净,然后自然晾干并研磨成粉末,得到蚕豆粉;
(2)取10g蚕豆粉置于聚四氟乙烯内衬中,然后添加50ml去离子水,再将聚四氟乙烯内衬置于超声清洗器中边超声,边用玻璃棒将蚕豆粉与去离子水搅拌混合均匀得到分散均匀的混合物。
(3)将上述聚四氟乙烯内衬密封于不锈钢反应釜中,拧紧不锈钢反应釜盖,放入190℃恒温干燥箱中反应12h后将反应釜取出,放于通风橱中使反应产物自然冷却至室温。
(4)将上述冷却后的反应产物置于离心管中,在11000r/min转速下离心15min;然后用移液枪将离心得到的上清液转移至截留分子量为500的透析袋中,并将透析袋置于去离子水中在室温条件下进行透析,同时用40r/min的速度缓慢搅拌透析袋,每间隔6h更换一次去离子水,共透析24h,最后收集透析袋内的液体得到碳点溶液。
(5)将碳点溶液进行冷冻干燥得到的褐色粉末,即为碳点(cds)。
实施例4
同实施例1的区别在于:步骤(2)中,在添加去离子水后,继续添加2.4g乙二胺,其它同实施例1,最后得到氮掺杂碳点(n-cds)。
实施例5
同实施例2的区别在于:步骤(2)中,在添加去离子水后,继续添加4g乙二胺,其它同实施例1,最后得到氮掺杂碳点(n-cds)。
实施例6
同实施例3的区别在于:步骤(2)中,在添加去离子水后,继续添加4.8g乙二胺,其它同实施例1,最后得到氮掺杂碳点(n-cds)。
对比例1
同实施例4的区别在于:去掉步骤(1),将蚕豆粉替换为纤维素,其它同实施例4。
对比例2
同实施例4的区别在于:去掉步骤(1),将蚕豆粉替换为草莓汁,其它同实施例4。
如图1所示为实施例1的cds的tem图,由该tem图可得cds的粒径为24~52nm之间(平均粒径为37nm);如图2所示为实施例4的n-cds的tem图,由该tem图可得n-cds的粒径为9~24nm之间(平均粒径为37nm)。由此可以得出,n-cds较cds的粒径更小。
如图3所示为实施例1的cds的紫外可见吸收光谱图,图中可知:其吸收带位于280nm附近和380nm附近,前者归因于c=c的π-π*跃迁,后者主要是c=o的n-π*跃迁引起的;如图4所示为实施例4的n-cds的紫外可见吸收光谱图,图中可知:其吸收带也位于280nm附近和380nm附近,与图3对比,两个吸收带的强度都明显增强,说明氮掺杂后其共轭程度增加。
如图5所示为实施例1的cds的荧光光谱图,图中可知:其最大激发波长为346nm,346nm激发时,最大发射波长为418nm;如图6所示为实施例4的n-cds的荧光光谱图,图中可知:通过氮掺杂后,其最大激发波长为400nm,400nm激发时,最大发射波长为473nm。
如图7所示为实施例4的n-cds在不同波长光激发下的荧光发射光谱图(激发波长由340nm至470nm,步长为10nm),图中可知:n-cds的发射峰随着激发波长的变大而发生红移。
如图8所示为实施例1的cds的红外光谱图,图中可知:1400~1500cm-1处的峰是由c-h的弯曲振动引起,1685cm-1处的峰来自于c=o的伸缩振动,2945cm-1处的峰由c-h的伸缩振动引起的,3100~3500cm-1这个区间中较宽的峰来自于o-h的伸缩振动;如图9所示为实施例4的n-cds的红外光谱图,图中可知:3430cm-1的强吸收峰证实了o-h键和n-h键共存,2964cm-1和1388cm-1的吸收峰代表了c-h键的特征吸收峰,1640cm-1处的吸收峰属于典型的c=o键振动吸收峰,1577cm-1处吸收峰为c=c键吸收峰,值得一提的是,1490cm-1处的吸收峰验证了n-h键的存在,1430cm-1对应于c-n键,这两个峰只存在于n-cds中,1027cm-1吸收峰是c-o键吸收峰。
如图10所示为实施例1的cds的xrd谱图,图中可知:在20.66°有一个比较宽的峰,晶面间距d为0.244nm,与石墨结构的(002)方向的层间距相符合;如图11所示为实施例4的n-cds的xrd谱图,图中可知:在20.22°有一个比较宽的峰,晶面间距d为0.248nm,与石墨结构的(002)方向的层间距相符合,n-cds的纵坐标最大值大于cds的,证明n-cds的含氮量多。
如图12所示为实施例1的cds的xps谱图,图中可知:在284.0ev、400.0ev和530.6ev处出现三个峰,分别对应c、n和o;如图13所示为实施例4的n-cds的xps谱图,图中可知:在284.0ev,400.0ev,530.6ev处出现三个峰,分别对应c、n和o,n-cds的400.0ev的吸收峰明显强于cds的,证明n-cds的含氮量明显多于cds的。
将上述实施例4得到的n-cds用于水样中重金属离子hg2+的测定:
1、n-cds对hg2+的测定灵敏度实验:在采用不同浓度hg2+离子滴定n-cds溶液的实验中,在紫外灯下可以观察到向n-cds溶液中加入hg2+溶液,发生了明显的荧光淬灭现象,通过逐渐增加hg2+浓度,获得了hg2+在(0~2)×10-4m浓度下的工作曲线;如图14所示为采用不同浓度的hg2+滴定n-cds的荧光猝灭曲线图,表明随着n-cds溶液中hg2+浓度的增加,其荧光强度呈梯级降低;图15所示为采用不同浓度hg2+滴定n-cds的荧光强度的峰值的拟合工作曲线,可见其呈线性分布,线性拟合程度良好。
2、n-cds对hg2+的测定灵选择性实验:首先用移液枪移取3ml配制的0.05g/l的碳点溶液于干燥比色皿中,在400nm激发波长下,检测该碳点溶液的荧光强度;然后在比色皿中依次加入3ml0.05g/l碳点溶液+3ul0.5mol/l金属离子溶液进行碳点-金属离子响应实验(其中金属离子分别采用fe2+、cu2+、co2+、zn2+、mg2+、ca2+、pb2+、ba2+、mn2+、cd2+),以及在比色皿中依次加入3ml0.05g/l碳点溶液+3ul0.1mol/l金属离子溶液+3ul0.5mol/lhg2+进行碳点-金属离子响应实验(其中金属离子分别采用fe2+、cu2+、co2+、zn2+、mg2+、ca2+、pb2+、ba2+、mn2+、cd2+)。如图16所示为不同金属离子对n-cds的选择性竞争图,结果表明:当氮掺杂碳点溶液中分别单独加入fe2+、cu2+、co2+、zn2+、mg2+、ca2+、pb2+、ba2+、mn2+、cd2+后,400nm处的荧光强度基本不变,荧光猝灭效率都小于20%;当氮掺杂碳点溶液中加入hg2+,400nm处的荧光强度明显降低,猝灭效率为79%,说明hg2+有能力猝灭氮掺杂碳点的荧光,而其他金属离子基本没有猝灭效果,说明荧光氮掺杂碳点对hg2+的选择性高;当氮掺杂碳点溶液中分别单独加入fe2+、cu2+、co2+、zn2+、mg2+、ca2+、pb2+、ba2+、mn2+、cd2+后,再加入hg2+,400nm处的荧光强度明显降低,猝灭效率大于52%,同时说明采用蚕豆作为碳源制备的n-cds对hg2+具有较强的专一性,只有fe2+和cu2+对hg2+有轻度干扰,其他金属离子干扰较弱。
综上表明采用蚕豆制备的n-cds对hg2+的测定灵敏度及选择性都非常高。
将上述实施例4得到的n-cds加入不同ph的缓冲液中,然后对n-cds的荧光强度进行扫描,如图17所示为n-cds的荧光强度与ph关系图,结果表明在ph值为2~10之间n-cds的荧光强度变化不大。
将上述实施例4得到的n-cds用于细胞成像研究:
1、采用mtt法分别用helacells检测n-cds的细胞毒性,如图18所示为n-cds的细胞毒性实验结果图,图中可知:在实验浓度(0.1mg/ml)下,细胞存活率超过95%,说明n-cds没有明显的毒性,并且与hela细胞具有细胞相容性,这为它们在体内和生物传感器中成像提供了一个有前景的候选。
2、为探讨合成的n-cds作为生物显像剂的可能性,选择hela细胞与n-cds共孵育,用共聚焦激光扫描显微镜观察hela细胞对n-cds的摄取情况,如图19为本发明实施例4制备的n-cds在hela细胞中的荧光成像图及n-cds在hela细胞中对hg2+的响应图。如图19a至19d所示,可以看出:hela细胞(无碳点)的亮场图像(如图19a所示)没有显示出可检测的发射信号;而用n-cds标记后,细胞在405nm激发波长下表现出较强的蓝色荧光(如图19b所示),在488nm激发波长下表现出较强的绿色荧光(如图19c所示),在559nm激发波长下表现出较强的红色荧光(如图19d所示)。上述结果表明,制备的n-cds可以替代有机染料或半导体量子点应用于生物成像。
3、研究hg2+是否能抑制n-cds染色hela细胞的荧光强度。如图19e所示为hela细胞加入hg2+的亮场图像;如hg2+与n-cds染色的hela细胞孵育30min后(如图19f至19h所示),与未加hg2+的n-cds染色的hela细胞(如图19a至19d所示)相比,蓝色(如图19f所示)、绿色(如图19g所示)、红色(如图19h所示)的荧光强度均明显下降。结果表明,采用蚕豆制备的n-cds在hela细胞内可作为hg2+荧光探针。
将本发明实施例4得到的n-cds与对比例1和对比例2得到的n-cds的量子产率进行比较;以及比较本发明实施例4得到的n-cds与对比例1和对比例2得到的n-cds用于水样中重金属离子hg2+的测定时的检测范围和检测限,结果如下表1所示。
表1
由上表1结果可知,相比其它生物质碳源纤维素和草莓汁,本发明采用蚕豆制备的n-cds具有较高的量子产率,同时具有较宽的检测范围及较低的检测限。
需要说明的是,上述实施例2和实施例3制备的cds同实施例1制备的cds的性能效果相当,上述实施例5和实施例6制备的n-cds同实施例4制备的cds的性能效果相当。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
此外,本领域的技术人员能够理解,尽管在此的一些实施例包括其它实施例中所包括的某些特征而不是其它特征,但是不同实施例的特征的组合意味着处于本发明的范围之内并且形成不同的实施例。例如,在上面的权利要求书中,所要求保护的实施例的任意之一都可以以任意的组合方式来使用。公开于该背景技术部分的信息仅仅旨在加深对本发明的总体背景技术的理解,而不应当被视为承认或以任何形式暗示该信息构成已为本领域技术人员所公知的现有技术。
- 还没有人留言评论。精彩留言会获得点赞!