表面改性金属化合物颗粒及表面改性金属化合物颗粒的制备方法与流程
本发明涉及一种表面改性金属化合物颗粒及表面改性金属化合物颗粒的制备方法。
背景技术:
金属氧化物颗粒及金属氢氧化物颗粒等金属化合物颗粒广泛应用在耐火物、陶瓷、光学材料以及汽车尾气催化剂等领域中。由于在这些用途中金属化合物颗粒多经由溶剂与其它材料混合使用,因此,需要一种根据用途可容易分散于合适的溶剂中的金属化合物颗粒。其中,对可分散于甲苯、正己烷、环己烷等非极性溶剂中的金属化合物颗粒的潜在需求很大。
一直以来,众所周知,为了使金属化合物颗粒分散于有机溶剂中而用羧酸等表面活性剂对金属化合物颗粒进行表面改性。
现有技术文献
专利文献
专利文献1:日本特开2008-31023号
专利文献2:日本特开2007-254257号
专利文献3:日本特开2006-82994号
技术实现要素:
要解决的技术问题
然而,一直以来并未公开一种表面活性剂的吸附状态的控制方法,以使金属化合物颗粒对有机溶剂具有良好的分散性。也就是说,一直以来并未发现优化了在甲苯、正己烷、环己烷等通常使用的非极性溶剂中的分散性的、由羧酸进行了表面改性的金属化合物颗粒。
本发明是鉴于上述问题而完成的,其目的在于提供一种对甲苯、正己烷、环己烷等非极性溶剂具有良好分散性的表面改性金属化合物颗粒及该表面改性金属化合物颗粒的制备方法。
用于解决问题的技术方案
发明人对表面改性金属化合物颗粒进行了深入研究。结果发现可以通过采用下述构成,使表面改性金属化合物颗粒对非极性溶剂具有良好的分散性,直至完成本发明。
即,本发明提供一种表面改性金属化合物颗粒,其中,
上述表面改性金属化合物颗粒包括通过第一羧酸和第二羧酸进行了表面改性的金属化合物颗粒,上述第一羧酸选自由甲基丙烯酸、丙烯酸和丙酸组成的群中的一种或多种,上述第二羧酸选自由碳数为6-16的脂肪酸和具有至少一个苯环的碳数为7-32的一价羧酸组成的群中的一种或多种,
上述第一羧酸的至少一部分是羧基的氢原子未被解离为离子的羧酸型。
上述第二羧酸(碳数为6-16的脂肪酸和具有至少一个苯环的碳数为7-32的一价羧酸)具有作为亲水基团的羧基(-cooh)、和占据第二羧酸相对较大部分的亲脂基团。在表面改性金属化合物颗粒中,大多数第二羧酸使羧基指向金属化合物颗粒的表面,使亲脂基团指向外侧。
根据上述构成,金属化合物颗粒通过上述第二羧酸进行表面改性,被占据第二羧酸相对较大部分的亲脂基团覆盖,因此对甲苯、正己烷、环己烷等非极性溶剂具有良好的分散性。
这里,若只是为了优化对非极性溶剂的分散性,则增加第二羧酸的含量即可。这是因为,只要增加第二羧酸的含量,就能够完全包围金属化合物颗粒。
然而,由于第二羧酸的亲脂基团部分的分子量较高,因此,若增加含量则会影响所得表面改性金属化合物颗粒的物理性质。因此,根据用途不同,有时可能会不允许这种物理性质的变化。具体地,例如,若增加第二羧酸的含量,则所得表面改性金属化合物颗粒的溶剂分散液的粘度会增高。
另一方面,若为了减小对所得表面改性金属化合物颗粒的物理性质的影响,而减少第二羧酸的含量,则对非极性溶剂的分散性会变差。对于其原因,发明人推测是因为,(1)包围金属化合物颗粒的亲脂基团的密度整体较为稀疏,而且,(2)一个亲脂基团和其他亲脂基团因相互作用紧密贴合在一起,金属化合物颗粒的表面主要分为被亲脂部分(多个亲脂基团紧密贴合在一起的部分)覆盖的部分和没有被覆盖的部分这两大部分。
因此,发明人发现,即使是在减少了第二羧酸的含量的情况下,只要以羧酸型的形式含有第一羧酸,就能够在不对表面改性金属化合物颗粒的物理性质带来较大影响的前提下,优化对非极性溶剂的分散性。
对于其原因,发明人推测是因为,如果以羧酸型的形式含有第一羧酸,则第一羧酸会进入第二羧酸的亲脂基团彼此之间,能够抑制第二羧酸的一个亲脂基团与其他亲脂基团紧密贴合在一起。即,推测即使第二羧酸的亲脂基团的密度整体比较稀疏,由于亲脂基团均匀包围金属化合物颗粒,也能够优化对非极性溶剂的分散性。进一步地推测,进入了第二羧酸的亲脂基团彼此之间的第一羧酸的亲脂基团能够优化对非极性溶剂的分散性。
此外,从实施例中明确的是,第一羧酸以羧酸型的形式存在。
优选的是,在上述构成中,上述第二羧酸为羧基的氢原子被解离成为离子状态的羧酸盐型。
若上述第二羧酸为羧基的氢原子被解离成为离子状态的羧酸盐型(-coo-),则能够适当地将上述第二羧酸吸附或者结合在金属化合物颗粒的表面上。
优选的是,在上述构成中,构成上述金属化合物颗粒的金属选自由锆和稀土类金属组成的群中的一种或多种。
若构成上述金属化合物颗粒的金属选自由锆和稀土类金属组成的群中的一种或多种,则在催化剂、电子零件、精细陶瓷、固体氧化物型燃料电池、以及光学等领域中的工业利用价值较高。
优选的是,在上述构成中,构成上述金属化合物颗粒的金属化合物为金属氧化物。
若构成上述金属化合物颗粒的金属化合物为金属氧化物,则在催化剂、电子零件、精细陶瓷、固体氧化物型燃料电池、以及光学等领域中的工业利用价值更高。
另外,本发明提供的表面改性金属化合物颗粒的制备方法是一种如上所述的表面改性金属化合物颗粒的制备方法,
包括:工序a,将第二羧酸添加到分散在水溶剂中的、zeta电位为正的金属化合物颗粒中,上述第二羧酸选自由碳数为6-16的脂肪酸和具有至少一个苯环的碳数为7-32的一价羧酸组成的群中的一种或多种;
工序b,对经上述工序a得到的产物进行干燥;以及
工序c,在上述工序b后,添加第一羧酸,所述第一羧酸选自由甲基丙烯酸、丙烯酸和丙酸组成的群中的一种或多种。
通过上述工序a、上述工序b、及上述工序,得到如上所述的表面改性金属化合物颗粒。即,根据上述构成,能够在不对表面改性金属化合物颗粒的物理性质带来较大影响的前提下,得到对非极性溶剂具有良好分散性的表面改性金属化合物颗粒。
优选的是,在上述构成中,上述工序b后的产物的金属化合物浓度以氧化物计为80重量%以下。
若上述工序b后的产物的金属化合物浓度以氧化物计为80重量%以下,则成为含有至少少量的水的状态。发明人推测,若将上述工序b后的产物的金属化合物浓度以氧化物计控制在80重量%以下,则能够形成水(水分子)存在于多个第二羧酸的亲脂基团彼此之间的状态。即,推测处于第二羧酸的亲脂基团彼此之间难以紧密贴合在一起的状态。然后,推测若在水存在于多个第二羧酸的亲脂基团彼此之间的状态下添加第一羧酸,则第一羧酸能够恰当地进入第二羧酸的亲脂基团彼此之间,能够抑制第二羧酸的一个亲脂基团和其他亲脂基团紧密贴合在一起。
由此,推测即使第二羧酸的亲脂基团的密度整体比较稀疏,由于亲脂基团均匀包围金属化合物颗粒,因此也能够优化对非极性溶剂的分散性。进一步地推测进入了第二羧酸的亲脂基团彼此之间的第一羧酸的亲脂基团能够优化对非极性溶剂的分散性。此外,尽管对此机制仅是推测,但从实施例可以明确的一点是:通过将上述工序b后的产物的金属化合物浓度以氧化物计设为80重量%以下,对非极性溶剂的分散性变得良好。
发明效果
根据本发明,能够优化表面改性金属化合物颗粒对非极性溶剂的分散性。
附图说明
图1是示出了用于对确定在表面改性金属化合物颗粒中第一羧酸以羧酸型的形式存在的方法进行说明的红外吸收光谱的图。
具体实施方式
以下,对本发明的实施方式进行说明。但本发明不仅限于这些实施方式。
<表面改性金属化合物颗粒>
本实施方式提供的表面改性金属化合物颗粒包括通过第一羧酸和第二羧酸进行了表面改性的金属化合物颗粒,上述第一羧酸选自由甲基丙烯酸、丙烯酸和丙酸组成的群中的一种或多种,上述第二羧酸选自由碳数为6-16的脂肪酸和具有至少一个苯环的碳数为7-32的一价羧酸组成的群中的一种或多种,上述第一羧酸的至少一部分是羧基的氢原子未被解离为离子的羧酸型。
上述第二羧酸(碳数为6-16的脂肪酸和具有至少一个苯环的碳数为7-32的一价羧酸)具有作为亲水基团的羧基(-cooh)、和占据第二羧酸相对较大部分的亲脂基团。在表面改性金属化合物颗粒中,大多数第二羧酸使羧基指向金属化合物颗粒的表面,亲脂基团指向外侧。
根据上述表面改性金属化合物颗粒,金属化合物颗粒通过上述第二羧酸进行表面改性,由于被占据第二羧酸相对较大部分的亲脂基团覆盖,因此对甲苯、正己烷、环己烷等非极性溶剂具有良好的分散性。
这里,若只是为了优化对非极性溶剂的分散性,则增加第二羧酸的含量即可。这是因为,只要增加第二羧酸的含量,就能够将金属化合物颗粒完全包围。
然而,由于第二羧酸的亲脂基团部分的分子量较高,因此,若增加含量会影响所得表面改性金属化合物颗粒的物理性质。因此,根据用途不同,有时可能会不允许这种物理性质的变化。具体来说,例如,若增加第二羧酸的含量,则所得表面改性金属化合物颗粒的粘度会增高。
另一方面,若为了减小对所得表面改性金属化合物颗粒的物理性质的影响,而减少第二羧酸的含量,则对非极性溶剂的分散性会变差。对于其原因,发明人推测是因为,(1)包围金属化合物颗粒的亲脂基团的密度整体较为稀疏,而且,(2)一个亲脂基团和其他亲脂基团因相互作用紧密贴合在一起,金属化合物颗粒的表面主要分为被亲脂部分(多个亲脂基团紧密贴合在一起的部分)覆盖的部分和没有被覆盖的部分这两大部分。
因此,发明人发现,即使在减少第二羧酸的含量的情况下,只要以羧酸型的形式含有第一羧酸,就能够在不对表面改性金属化合物颗粒的物理性质带来较大影响的前提下,优化对非极性溶剂的分散性。
对于其原因,发明人推测是因为,如果以羧酸型的形式含有第一羧酸,第一羧酸会进入第二羧酸的亲脂基团彼此之间,则能够抑制第二羧酸的一个亲脂基团和其他亲脂基团紧密贴合在一起。即,推测即使第二羧酸的亲脂基团的密度整体比较稀疏,由于亲脂基团均匀包围金属化合物颗粒,也能够优化对非极性溶剂的分散性。进一步地推测,进入了第二羧酸的亲脂基团彼此之间的第一羧酸的亲脂基团,能够优化对非极性溶剂的分散性。
此外,从实施例中明确的是,第一羧酸以羧酸型的形式存在。
在上述表面改性金属化合物颗粒分散于非极性溶剂时,以羧酸型的形式吸附在金属化合物颗粒上的第一羧酸是通过何种机制有助于溶剂分散尚不明确,作为假说认为,通过羧酸型的第一羧酸取代溶剂分子,促进了表面改性金属化合物颗粒的溶剂化。由于即使使用醋酸、甲酸、乙酸乙酯等代替第一羧酸(甲基丙烯酸、丙烯酸、丙酸),也无法得到分散效果,因此认为第一羧酸的分子量、偶极矩、极化率等物理性质适合于该作用机制。此外,第二羧酸即使以羧酸型的形式存在也没关系,但或许是因为分子量等不适合,发挥不出如第一羧酸那样的功能。
作为构成上述金属化合物颗粒的金属化合物,可以列举金属氧化物、金属氢氧化物等。其中,从工业利用价值的观点来看,优选为金属氧化物。在本说明书中,“金属氧化物”、“金属氢氧化物”是指,以金属原子、氧原子及羟基为基本构成要素的所有物质。例如,也包含这些的混合物、复合物及固溶体。另外,也包括包含杂质(例如,不可避免的杂质)的情况。
上述金属氧化物及上述金属氢氧化物的种类没有特别限制,可以列举:经常作为颗粒使用的氧化锆、二氧化铪、二氧化铈、其他稀土类氧化物;二氧化钛、氧化铝、二氧化硅、氢氧化锆、氢氧化铪、氢氧化铈、其他稀土类氢氧化物;氢氧化钛、氢氧化铝;这些化合物的混合物、这些化合物的复合物及这些化合物的固溶体等。其中,从工业利用价值的观点来看,构成上述金属化合物颗粒(例如,上述金属氧化物、上述金属氢氧化物)的金属优选选自由锆和稀土类金属组成的群中的一种或多种。
上述金属氧化物及上述金属氢氧化物不限定结晶状态(晶体或非晶体)及晶体体系的种类。
锆化合物作为不可避免的杂质,通常含有铪。具体地,以氧化物计,锆化合物含有铪的量,通常为用下式(1)计算出的氧化铪的含量,为1.3-2.5重量%左右。
<式(1)>
([氧化铪的重量]/([氧化锆的重量]+[氧化铪的重量]))×100(%)
另外,氧化锆作为结晶相稳定剂,也可以含有稀土类元素、碱金属元素及碱土类金属元素。另外,为了改变上述金属氧化物、上述金属氢氧化物的催化特性、光催化特性、荧光特性以及吸光特性等物理性质等,也可以添加除金属元素以外的元素。另外,只要在作为表面改性金属化合物颗粒使用时不对其造成不良影响,也可以含有除上述以外的杂质。
上述金属化合物颗粒通过从由甲基丙烯酸、丙烯酸及丙酸组成的群中选择的一种以上的第一羧酸、和从由碳数为6-16的脂肪酸和具有至少一个苯环的碳数为7-32的一价羧酸组成的群中选择的一种以上的第二羧酸,进行表面改性。
“进行表面改性”是指,包括(1)上述第一羧酸、上述第二羧酸化学或者物理地吸附在金属化合物颗粒的表面上的状态;及(2)上述第一羧酸进入上述第二羧酸的亲脂基团彼此之间的状态。
在上述(1)的情况下,上述第一羧酸及上述第二羧酸基本上是以羧基的氢原子被解离成为离子状态的羧酸盐型的形式吸附在金属化合物颗粒的表面上。
在上述(2)的情况下,上述第一羧酸是以羧基的氢原子未被解离为离子的羧酸型的形式进入上述第二羧酸的亲脂基团彼此之间。此外,在该情况下,上述第一羧酸不是化学或者物理地吸附在金属化合物颗粒的表面上的状态。
如上所述,上述第二羧酸选自由碳数为6-16的脂肪酸和具有至少一个苯环的碳数为7-32的一价羧酸组成的群中的一种或多种。
在本说明书中,碳数为6-16的脂肪酸是指,包括羧基中包含的碳原子,分子中具有6-16个碳原子的一价羧酸。具体地,例如庚酸[ch3(ch2)5cooh]是碳数为7的脂肪酸。
上述碳数为6-16的脂肪酸可以是直链型、侧链型中的任意一种。另外,分子中也可以具有双键。另外,也可以具有环状结构。从功能性、经济性的观点来看,上述碳数为6-16的脂肪酸优选为碳数为7-12的直链型脂肪酸。具体地,优选为庚酸、辛酸、壬酸、癸酸、十二酸。
另外,在本说明书中,具有至少一个苯环的碳数为7-32的一价羧酸没有特别限制,具体地,例如,可以列举(a)具有一个苯环的碳数为7-32的一价羧酸;(b)具有两个苯环的碳数为14-32的一价羧酸。
具体地,例如,3-苯基丙酸[c6h5ch2ch2cooh]为具有一个苯环的碳数为9的一价羧酸。
作为上述(a),没有特别限制,例如可以列举下述(a-1)-(a-6)。
(a-1)安息香酸[c6h5cooh]
(a-2)r1-ph-cooh
(其中,式中的ph为苯环,r1为烃,苯环和r1的碳数的总和在7-31的范围内。)
(a-3)ph-r2-cooh
(其中,式中ph为苯环,r2为烃,苯环和r2的碳数的总和在7-31的范围内。)
(a-4)r3-ph-r4-cooh
(其中,式中ph为苯环,r3及r4为烃,苯环和r3、r4的碳数的总和在8-31的范围内。)
(a-5)r5-ph(me)x-r6-cooh
(其中,式中ph为苯环,me为甲基,x为1-4的整数,r5及r6为烃,苯环、甲基、r5以及r6的碳数的总和在9-31的范围内。)
(a-6)r7-ph(et)x-r8-cooh
(其中,式中ph为苯环,et为乙基,x为1-4的整数,r7及r8为烃,苯环、乙基、r7以及r8的碳数的总和在10-31的范围内。)
上述用r1-r8表示的烃可以是直链型,也可以具有支链。另外,可以具有双键,也可以具有环状结构。
作为上述(b),没有特别限制,例如可以列举下述(b-1)。
(b-1)r9-biph-r10-cooh
(其中,式中biph为联苯结构,r9及r10为烃,联苯结构和r9、r10的碳数的总和在14-31的范围内。)
上述用r9、r10表示的烃可以是直链型,也可以具有支链。另外,可以具有双键,也可以具有环状结构。
如上所述,优选的是,上述第二羧酸以羧基的氢原子被解离成为离子状态的羧酸盐型的形式吸附在上述金属化合物颗粒的表面上。但是,不一定必须上述第二羧酸全部是以羧酸盐型的形式吸附在上述金属化合物颗粒的表面上,至少一部分,优选大部分以羧酸盐型的形式吸附在上述金属化合物颗粒的表面上即可。
如上所述,上述第一羧酸选自由甲基丙烯酸、丙烯酸和丙酸组成的群中的一种或多种,上述第一羧酸的至少一部分是羧基的氢原子未被解离为离子的羧酸型。
关于确定上述第一羧酸以羧酸型的形式存在的方法,后面会进行描述。
在上述表面改性金属化合物颗粒分散在甲苯中时,中值粒径优选为1-100nm,更优选为1-50nm,进一步优选为1-30nm。若将上述表面改性金属化合物颗粒分散在甲苯中时的中值粒径为1-100nm,则对非极性溶剂的分散性会更好。
此外,上述中定义了分散在甲苯中时的中值粒径是因为,若为了定义中值粒径,分散在作为非极性溶剂的代表的甲苯中时的中值粒径为1-100nm,则即使是除甲苯以外的非极性溶剂,也可以与甲苯相同,优化分散性。因此,分散在甲苯中时的中值粒径为1-100nm,不仅仅是对甲苯,也是用来表示对除甲苯以外的非极性溶剂的分散性更好的指标,是指对除本甲苯以外的非极性溶剂具有良好的分散性。
这里,中值粒径是指,在通过动态光散射进行的粒度测量中,颗粒的累积体积频率为50%的粒径。
作为能够分散表面改性金属化合物颗粒的非极性溶剂,没有特别限制。上述非极性溶剂的具体示例可以列举:苯、甲苯、二甲苯、正己烷、环己烷、石油醚、以及矿油精等。此外,上述石油醚是指石油的低沸点馏分,不含有作为化学物质的醚。
以金属氧化物计,上述表面改性金属化合物颗粒相对甲苯优选溶解或者分散60重量%以上,更优选溶解或者分散65重量%以上。此外,溶解或者分散是指,后述透光率在后述数值范围内。
对应于使用该表面改性金属化合物颗粒的产品的特性等不同,上述表面改性金属化合物颗粒的粘度所优选的值也不同,例如,优选在0.1-10000mpa·s的范围内,更优选在21-1000mpa·s的范围内。上述粘度取决于实施例中所述的方法。
以氧化物计,上述表面改性金属化合物颗粒以60重量%溶解或分散在甲苯中时的透光率优选在波长400nm时为1%以上且在波长800nm时为50%以上,更优选的是在波长400nm时为5%以上且在波长800nm时为60%以上。若上述透光率在波长400nm时为1%以上且在波长800nm时为50%以上,则可以说上述表面改性金属化合物颗粒恰当地分散在非极性溶剂中。此外,在将上述表面改性金属化合物颗粒溶解或者分散在甲苯中时,在观察到有沉淀的情况下,即使上述透光率满足上述数值范围,也不能说恰当地分散在非极性溶剂中。
<表面改性金属化合物颗粒的制备方法>
本实施方式提供的表面改性金属化合物颗粒的制备方法至少如下工序:
工序a,将第二羧酸添加到分散在水溶剂中的、zeta电位为正的金属化合物颗粒中,上述第二羧酸选自由碳数为6-16的脂肪酸和具有至少一个苯环的碳数为7-32的一价羧酸组成的群中的一种或多种;
工序b,对经上述工序a得到的产物进行干燥;以及
工序c,在上述工序b后,添加第一羧酸,所述第一羧酸选自由甲基丙烯酸、丙烯酸和丙酸组成的群中的一种或多种。
更优选的是,本实施方式提供的表面改性金属化合物颗粒的制备方法至少包括如下工序:
工序a,将第二羧酸添加到分散在水溶剂中的、zeta电位为正的金属化合物颗粒中,上述第二羧酸选自由碳数为6-16的脂肪酸和具有至少一个苯环的碳数为7-32的一价羧酸组成的群中的一种或多种;
工序x,将第一羧酸添加到分散在水溶剂中的、zeta电位为正的金属化合物颗粒中,上述第一羧酸选自由甲基丙烯酸、丙烯酸和丙酸组成的群中的一种或多种;
工序y,用纯净水清洗经上述工序a及上述工序x得到的产物;
工序b,对经上述工序a、上述工序x、及上述工序y得到的产物进行干燥;以及
工序c,在上述工序b后,添加第一羧酸,所述第一羧酸选自由甲基丙烯酸、丙烯酸和丙酸组成的群中的一种或多种。
下面对各个工序进行说明。此外,在下面说明的各个工序中,通过根据一般的化学反应速度的原理调整各个工序中的温度等,可以根据制备的便利性适当地改变每个工序中的反应速率。
<工序a及工序x>
首先,准备分散在水溶剂中的金属化合物颗粒。金属化合物颗粒可以列举上述的金属化合物颗粒。作为分散在水溶剂中的金属化合物颗粒,准备金属化合物颗粒(例如,金属氧化物颗粒、金属氢氧化物颗粒或金属氧化物颗粒与金属氢氧化物颗粒的混合物)以1-100nm的中值粒径分散在水中的溶胶。构成上述金属氧化物颗粒、金属氢氧化物颗粒的金属没有特别限制,例如可以列举锆、铈、其它稀土类、钛、铝、硅等。分散在水溶剂中的金属化合物颗粒的具体示例例如可以列举专利文献1、专利文献2、专利文献3中公开的氧化锆溶胶或二氧化铈溶胶。
下面,用作起始原料、分散在水溶剂中的金属化合物颗粒也可以称为被改性颗粒。
上述被改性颗粒的zeta电位优选为正。这是因为,在后段的表面改性处理中,由于zeta电位为正,带负电荷的第二羧酸的羧酸盐(coo-)被吸引到上述被改性颗粒的表面并吸附或结合,由此,对非极性溶剂具有较高亲和性的亲脂基团(脂肪链)指向上述被改性颗粒的外侧,从而形成适于对非极性溶剂的溶剂化的取向状态。
作为向上述被改性颗粒中添加第二羧酸的方法(工序a),例如,在搅拌上述被改性颗粒(例如,溶胶)时,添加作为酒精溶液的第二羧酸。搅拌时间没有特别限制,例如,可以是30分钟-2小时。
上述酒精溶液中使用的酒精例如可以使用甲醇、乙醇、正丙醇、异丙醇、正丁醇、苯甲醇等。此外,即使是除酒精以外的溶剂,只要能够溶解上述第二羧酸就可以使用。
上述工序a中的上述第二羧酸的浓度没有特别限制,优选为1-30重量%,更优选为5-25重量%。通过使上述工序a中的上述第二羧酸的浓度在上述数值范围内,能够将向金属化合物颗粒的吸附反应控制在合适的速度。
在上述工序a中,上述第二羧酸和构成上述金属化合物颗粒的金属的摩尔比([第二羧酸的摩尔数]/[构成金属化合物颗粒的金属的摩尔数])优选在0.01-0.2的范围内,更优选在0.02-0.15的范围内。通过使上述工序a中的上述摩尔比在上述数值范围内,能够抑制对所得表面改性金属化合物颗粒的物理性质(例如,粘度)带来较大影响。
尤其是在构成上述金属化合物颗粒的金属是锆时,在上述工序a中,上述第二羧酸与锆的摩尔比([第二羧酸的摩尔数]/[锆])优选在0.01-0.2的范围内,更优选在0.02-0.15的范围内。
在上述工序a后,根据需要进行工序x。具体地,在工序x中,例如,在搅拌工序a中得到的分散溶液(添加了第二羧酸的被改性颗粒)时,添加未稀释的或者用与上述相同的酒精等稀释的第一羧酸。搅拌时间没有特别限制,例如可以设为30分钟-2小时。
工序x是在第二羧酸对金属化合物颗粒表面的疏水化不充分时进行的工序,在金属化合物颗粒表面的疏水化充分时也可以不进行工序x。但是,若在工序x中增加第二羧酸的添加量,会对所得表面改性金属化合物颗粒的物理性质带来较大影响,因此,优选的是,通过减少分子量大的第二羧酸的添加量,添加分子量小的第一羧酸,在不对所得表面改性金属化合物颗粒的物理性质带来较大影响的前提下,弥补疏水化。
此外,在工序x中添加的第一羧酸以羧基的氢原子被解离成为离子状态的羧酸盐型的形式吸附在金属化合物颗粒表面。即,在工序a及工序x后,第一羧酸及第二羧酸使羧基指向金属化合物颗粒的表面以羧酸盐型的形式吸附。
另外,在进行工序x时,由于存在羧酸盐型的第一羧酸,因此能够抑制多个第二羧酸的亲脂基团紧密贴合在一起。
在上述工序x中,上述第一羧酸与构成上述金属化合物颗粒的金属的摩尔比([第一羧酸的摩尔数]/[构成金属化合物颗粒的金属的摩尔数])优选在0.01-0.5的范围内,更优选在0.1-0.4的范围内。通过使上述工序x中的上述摩尔比在上述数值范围内,能够在吸附于金属化合物颗粒的表面上的多个第二羧酸彼此之间恰当地配置第一羧酸。
尤其是在构成上述金属化合物颗粒的金属为锆时,在上述工序x中,上述第二羧酸与锆的摩尔比([第一羧酸的摩尔数]/[锆])优选在0.01-0.5的范围内,更优选在0.1-0.4的范围内。
经过上面的工序a及工序x,通常可得到糊状的沉淀产物。
<工序y>
接下来,根据需要,用纯净水清洗经上述工序a及上述工序x得到的产物。具体地,将经过上述工序a及上述工序x得到的产物倒入纯净水中,或者在上述产物中加入纯净水等,去除附着在产物上的杂质。作为上述杂质,例如可以列举来自上述被改性颗粒原料的离子成分、在上述工序a和上述工序x中使用的酒精溶剂等。但是,在使用所得表面改性金属化合物颗粒及该表面改性金属化合物颗粒的产品中,上述杂质不成问题时,也可以不进行工序y。但是,通常,优选尽可能地去除上述杂质。
另外,通过工序y,吸附在金属化合物颗粒表面上的羧酸盐型形式的第一羧酸几乎会被全部冲洗掉。
<工序b>
接下来,干燥经上述工序a、上述工序x、及上述工序y得到的产物。干燥的方法没有特别限制,可以采用过滤、加热及这些方式的组合等。
干燥后的产物的金属化合物浓度、即上述工序b后的产物的金属化合物浓度以氧化物计优选在80重量%以下,更优选以氧化物计在75重量%以下。
若上述工序b后的产物的金属化合物浓度以氧化物计在80重量%以下,则成为含有至少少量的水的状态。发明人推测,若以氧化物计将上述工序b后的产物的金属化合物浓度控制在80重量%以下,则能够形成水(水分子)存在于多个第二羧酸的亲脂基团彼此之间的状态。即,推测处于第二羧酸的亲脂基团彼此难以紧密贴合在一起的状态。然后推测,若在水存在于多个第二羧酸的亲脂基团彼此之间的状态下添加第一羧酸,则第一羧酸能够恰当地进入第二羧酸的亲脂基团彼此之间,能够抑制第二羧酸的一个亲脂基团与其他亲脂基团紧密贴合在一起。
由此推测,即使第二羧酸的亲脂基团的密度整体比较稀疏,由于亲脂基团均匀包围金属化合物颗粒,因此也能够优化对非极性溶剂的分散性。进一步地推测,进入了第二羧酸的亲脂基团彼此之间的第一羧酸的亲脂基团也能够优化对非极性溶剂的分散性。此外,尽管对此机制仅是推测,但从实施例可以明确:通过将上述工序b后的产物的金属化合物浓度设为80重量%以下,对非极性溶剂的分散性变得良好。
此外,上述工序b后的产物的金属化合物浓度的下限值没有特别限制。这是因为,在上述金属化合物浓度较低的情况下,通过增加在下一个工序c中添加的第一羧酸的量,便可得到期望的表面改性金属化合物颗粒。从制备的观点来看,以氧化物计,上述金属化合物浓度的下限值例如设为50重量%以上、60重量%以上等即可。
经上面的工序,得到前体粉末。
<工序c>
在上述工序b后,在上述前体粉末中添加第一羧酸。具体地,例如,不用溶剂等进行稀释,而将第一羧酸直接添加到上述前体粉末中混合。混合方法没有特别限制,可以举出例如混炼等。由此,存在于多个第二羧酸的亲脂基团彼此之间的水(水分子)通过与第一羧酸进行取代等,第一羧酸进入第二羧酸的亲脂基团彼此之间。从而能够抑制第二羧酸的一个亲脂基团与其他亲脂基团紧密贴合在一起。进一步地,进入了第二羧酸的亲脂基团彼此之间的第一羧酸的亲脂基团也能够优化对非极性溶剂的分散性。
此外,工序c中添加的第一羧酸以羧基的氢原子未被解离为离子的羧酸型的形式进入第二羧酸的亲脂基团彼此之间。即,在工序c后,第一羧酸以羧酸型的形式进入第二羧酸的亲脂基团彼此之间。由此,即使第二羧酸的亲脂基团的密度整体比较稀疏,由于亲脂基团均匀包围金属化合物颗粒,因此也能够优化对非极性溶剂的分散性。进一步地,进入了第二羧酸的亲脂基团彼此之间的第一羧酸的亲脂基团也能够优化对非极性溶剂的分散性。
在上述工序c中,上述第一羧酸与构成上述金属化合物颗粒的金属的摩尔比([第一羧酸的摩尔数]/[构成金属化合物颗粒的金属的摩尔数])优选在0.01-0.5的范围内,更优选在0.1-0.4的范围内。通过使上述工序c中的上述摩尔比在上述数值范围内,能够在第二羧酸的亲脂基团彼此之间恰当地配置第一羧酸。
尤其是,在构成上述金属化合物颗粒的金属是锆时,在上述工序c中,上述第一羧酸与锆的摩尔比([第一羧酸的摩尔数]/[锆])优选在0.01-0.5的范围内,更优选在0.1-0.4的范围内。
在工序c后,也可以根据需要进行干燥。
经过上面的工序,通常可得到粉末状的表面改性金属化合物颗粒。
上面对本实施方式提供的表面改性金属化合物颗粒的制备方法进行了说明。
最后,对确定在本实施方式提供的表面改性金属化合物颗粒中第一羧酸以羧酸型的形式存在的方法进行说明。
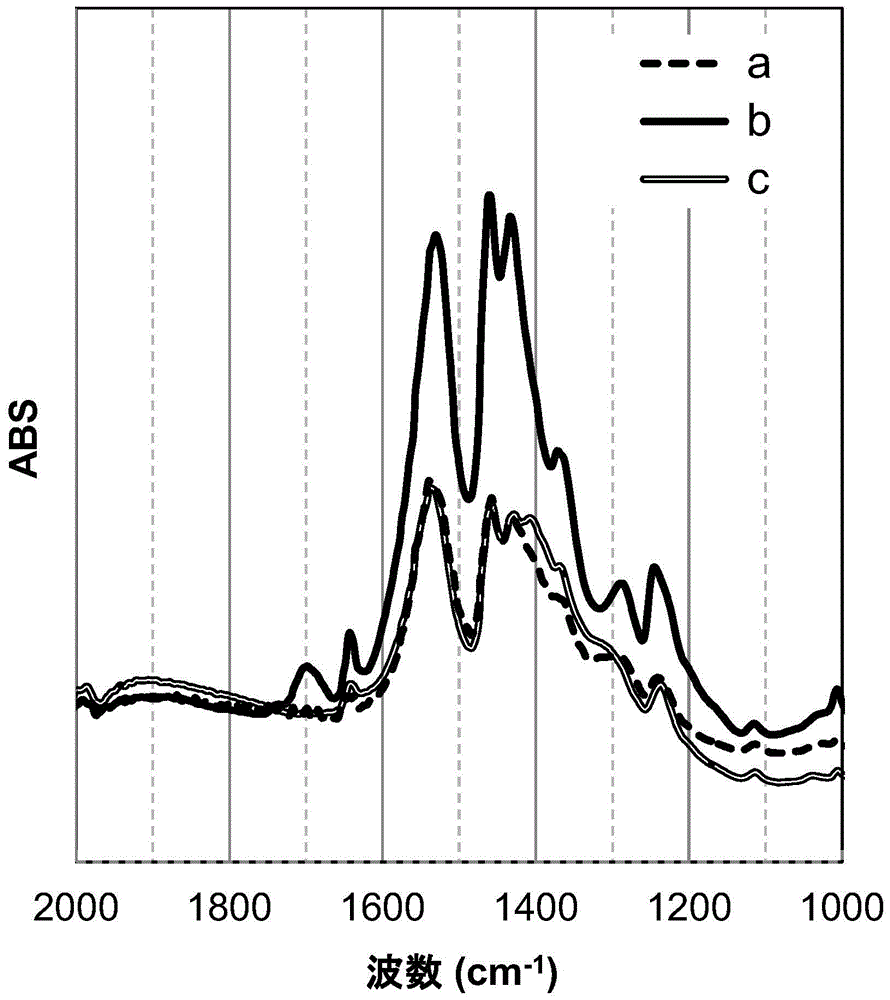
图1示出了用于对确定在表面改性金属化合物颗粒中第一羧酸以羧酸型的形式存在的方法进行说明的红外吸收光谱的图。
在图1中,a是在上述工序b后,上述工序c前的前体粉末(干燥后)的红外吸收光谱的一个示例。b是在上述工序c后所得表面改性金属化合物颗粒的红外吸收光谱的一个示例。c是用纯净水清洗所得表面改性金属化合物颗粒得到的粉末的红外吸收光谱的一个示例。此外,a是后述比较例1提供的表面改性氧化锆颗粒的红外吸收光谱,b是后述实施例1提供的表面改性氧化锆颗粒的红外吸收光谱,c是用纯净水清洗实施例1提供的表面改性氧化锆颗粒得到的粉末的红外吸收光谱。
在图1中,对齐显示基准线,以便于对各光谱进行对比。
首先,对光谱a和上述工序b后的状态进行比较。
如上所述,在上述工序b后,吸附在金属化合物颗粒表面上的羧酸盐型的第一羧酸几乎被全部冲洗掉。因此,在上述工序b后,是这样一种状态:羧酸中,只有羧酸盐型的第二羧酸吸附在金属化合物颗粒的表面上。图1的光谱a是在该状态,即只有羧酸盐型的第二羧酸吸附在金属化合物颗粒表面的状态下的光谱。
这里,在光谱a中,在1680-1720cm-1的区域中不存在属于cooh的c=o拉伸振动的峰值(以下也称为“1700cm-1的峰值”),而在1540-1580cm-1的区域中存在属于coo-的不对称拉伸振动的峰值(以下也称为“1560cm-1的峰值”)。这就意味着不存在羧酸型的羧酸,只存在羧酸盐型的羧酸。
即,光谱a符合上述工序b后的第二羧酸不是作为羧酸型存在,而是作为羧酸盐型存在。
接下来,对光谱b和上述工序c后的状态进行比较。
如上所述,上述工序c是在上述工序b后,添加第一羧酸的工序。因此,在上述工序c后,是在上述工序b后的状态下添加了第一羧酸的状态。而且如上所述,上述工序c后,第一羧酸的至少一部分以羧基的氢原子未被解离为离子的羧酸型的形式存在。
图1的光谱b是该状态,即羧酸盐型的第二羧酸吸附在金属化合物颗粒表面,且第一羧酸的至少一部分以羧酸型的形式存在的状态下的光谱。
这里,在光谱b中,存在1720cm-1的峰值。进一步地,与光谱a相比1560cm-1的峰值较大。
这就意味着,添加的第一羧酸的至少一部分作为羧酸型存在,且剩余的第一羧酸作为羧酸盐型存在。
即,光谱b符合上述工序c后的状态,即在本实施方式提供的表面改性金属化合物颗粒中,第一羧酸的至少一部分以羧酸型的形式存在。
接下来,比较光谱c和用纯净水清洗表面改性金属化合物颗粒后的状态。
用纯净水充分清洗表面改性金属化合物颗粒后,吸附在金属化合物颗粒的表面上的羧酸盐型的第一羧酸、和存在于多个第二羧酸的亲脂基团彼此之间的羧酸型的第一羧酸这两者会洗脱。
图1的光谱c是该状态,即羧酸盐型的第一羧酸、和羧酸型的第一羧酸这两者洗脱,只有羧酸盐型的第二羧酸吸附在金属化合物颗粒表面的状态下的光谱。
这里,光谱c称为与光谱a几乎相同的光谱。这就意味着是这样一种状态:羧酸盐型的第一羧酸、和羧酸型的第一羧酸这两者洗脱,与光谱a相同,只有羧酸盐型的第二羧酸吸附在金属化合物颗粒表面。
如上所述,通过在表面改性金属化合物颗粒的制备工序的各个工序中对比红外吸收光谱,能够确定在所得表面改性金属化合物颗粒中,将金属化合物颗粒的表面改性的第一羧酸的至少一部分是羧基的氢原子未被解离为离子的羧酸型。
另外,通过对比得到的表面改性金属化合物颗粒的红外吸收光谱和用纯净水充分清洗该表面改性金属化合物颗粒后的红外吸收光谱,能够确认表面改性金属化合物颗粒中是否存在羧酸型的第一羧酸。
实施例
下面,将示例性地对本发明优选的实施例进行详细说明。但是,除非另有特别的限制说明,本实施例中所述的材料和配合量等不旨在将本发明的范围限制于此。下面,重量%仅记为%。此外,在实施例及比较例中得到的表面改性氧化锆颗粒中,氧化铪作为不可避免的杂质相对氧化锆含有1.3-2.5重量%(通过下式(1)算出)。
<式(1)>
([氧化铪的重量]/([氧化锆的重量]+[氧化铪的重量]))×100(%)
实施例中使用的试剂如下所示。下面重量%仅记为%。
<试剂>
庚酸(东京化成工业)
癸酸(东京化成工业)
十二酸(东京化成工业)
3-苯基丙酸(东京化成工业)
戊酸(东京化成工业)
12-羟基硬脂酸(東京化成)
正丁醇(日本西格玛奥德里奇,1级)
甲基丙烯酸(东京化成工业)
丙烯酸(日本西格玛奥德里奇,1级)
丙酸(日本西格玛奥德里奇,1级)
88%甲酸(kishida化学工业,特级)
冰醋酸(日本西格玛奥德里奇,1级)
甲苯(日本西格玛奥德里奇,1级)
无水柠檬酸(kishida化学,特级)
25%氨水(日本西格玛奥德里奇,特级)
分析(测量)时使用的装置及测量条件如下所示。
<zeta电位测量>
装置:zetasizernanozs(马尔文)
测量条件:
氧化物浓度:30%
材料:
zro2(氧化锆溶胶)
ri:2.17
吸收率:0.01
ceo2(二氧化铈溶胶)
ri:2.15
吸收率:0.01
分散剂:水
温度:25℃
样品池:dts1060c
测量时间:自动(10-100次)
f(κa)选项:
型号:smoluchowski
自动衰减选项:yes
自动电压选项:yes
分析模型:自动模式
<ph测量>
装置:ph计d-51(堀场制作所制造),grt复合电极9681s-10d(堀场制作所制造)
测量条件:
zro2浓度:30%
温度:20-25℃
搅拌时测量。
<金属氧化物浓度的测量>
装置:
电炉:nhk-170(日陶科学)
坩埚:c1(nikkato)
测量条件:
在坩埚内称取样品1g,将在1000℃下烧制1小时后的样品的剩余重量视为金属氧化物重量。用精密天平测量重量,精确到1/10000g。
[实施例、比较例提供的表面改性金属化合物颗粒的制作]
(实施例1)
首先,通过以下步骤得到被改性颗粒(分散在水溶剂中的金属化合物颗粒),即氧化锆溶胶。
向886.7g氯氧化锆水溶液(以zro2计含有180.0gzr)中添加纯净水,制成1000g。另一方面,向747.9g氢氧化钠水溶液(以100%naoh的形式含有187.0gnaoh)中添加纯净水,制成1400g,加热到90℃。然后,加热到90℃的氢氧化钠水溶液中添加制得的氯氧化锆水溶液,其后,冷却至室温(25℃)。此时,溶液的ph为13.7。将该溶液过滤,用5000g的纯净水进行清洗,去除得到的氢氧化锆中的杂质,得到651.7g湿滤饼。从该湿滤饼中称取538.8g,放入烧杯中,添加纯净水,制成1000g。将此溶液搅拌10分钟,使氢氧化锆均匀分散。其后,添加60.35g的61%硝酸作为胶溶剂,加热至100℃,通过搅拌并保持72小时,得到氧化锆溶胶。
得到的溶液为透明的浅蓝色,判断其已经完全成为氧化锆溶胶。其后,冷却至室温后,用膜过滤器进行超滤,用500g的纯净水清洗2次,加入纯净水得到500.0g氧化锆溶胶。得到的氧化锆溶胶的ph为3.3,zro2浓度为30%,zeta电位为正。
搅拌该150g溶胶,并向其中添加23g(摩尔比[癸酸]/[zr]=0.073)癸酸(碳数为10的直链型脂肪酸)的正丁醇溶液(浓度:20%),搅拌30分钟(相当于工序a)。
接下来,在继续搅拌的同时添加4g(摩尔比[甲基丙烯酸]/[zr]=0.13)甲基丙烯酸,搅拌30分钟(相当于工序x)。停止搅拌后静置,得到透明的上清液和糊状的沉淀物。
搅拌1l的纯净水(去离子水)时滴下该沉淀物,得到粉末状沉淀。用布氏漏斗过滤该粉末状沉淀,再通入1l的纯净水进行清洗(相当于工序y)。
接下来,进行脱水、干燥,得到前体粉末(相当于工序b)。此时,进行干燥,使前体粉末的金属化合物浓度为,zro2浓度:70%。
接下来,在该前体粉末中添加摩尔比[甲基丙烯酸]/[zr]=0.13当量的甲基丙烯酸,充分混合(相当于工序c)。其后,在80℃的干燥机内干燥1小时,得到表面改性氧化锆颗粒。
(实施例2)
除在前体粉末中添加摩尔比[丙烯酸]/[zr]=0.13当量的丙烯酸以外,与实施例1相同,得到表面改性氧化锆颗粒。
(实施例3)
除在前体粉末中添加摩尔比[丙酸]/[zr]=0.13当量的丙酸以外,与实施例1相同,得到表面改性氧化锆颗粒。
(实施例4)
除代替癸酸的正丁醇溶液(浓度:20%),添加28g(摩尔比[庚酸]/[zr]=0.12)庚酸(碳数为7的直链型脂肪酸)的正丁醇溶液(浓度:20%)以外,与实施例1相同,得到表面改性氧化锆颗粒。
(实施例5)
除代替癸酸的正丁醇溶液(浓度:20%),添加25g(摩尔比[十二酸]/[zr]=0.068)十二酸(碳数为12的直链型脂肪酸)的正丁醇溶液(浓度:20%)以外,与实施例1相同,得到表面改性氧化锆颗粒。
(实施例6)
除代替癸酸的正丁醇溶液(浓度:20%),添加30g(摩尔比[3-苯基丙酸]/[zr]=0.082)3-苯基丙酸的正丁醇溶液(浓度:15%)以外,与实施例1相同,得到表面改性氧化锆颗粒。
(实施例7)
首先,通过以下的步骤得到被改性颗粒,即二氧化铈溶胶。
在回流下搅拌2000g含有硝酸铈的水溶液(以ceo2计含有120gce,ceo2浓度为6重量%;ph≦1),并在100℃下保持24小时。其后,在20-25℃的环境温度下静置一昼夜后,倾析去除上清液,留下沉淀物(二氧化铈溶胶前体),向其中加入硝酸铈和纯净水,制成2000g(不考虑该沉淀物的存在,以ceo2计含有120gce,ceo2浓度为6重量%,ph≦1)。在回流下再次搅拌使该沉淀物共存的含有硝酸铈的水溶液,并在1000℃下保持24小时。其后,在20-25℃的环境温度下静置一昼夜后,倾析去除上清液,留下沉淀物,向其中加入硝酸铈和纯净水,制成2000g(不考虑该沉淀物的存在,以ceo2的形式含有120gce,ceo2浓度6重量%,ph≦1)。在回流下再次搅拌使该沉淀物共存的含有硝酸铈的水溶液,并在100℃下保持24小时。其后,在20-25℃的环境温度下静置一昼夜后,倾析去除上清液,进行过滤。通过在得到的188g(湿)沉淀物中加入533ml纯净水,得到二氧化铈溶胶。
接下来,通过超滤进行纯化及浓缩,得到二氧化铈溶胶(ph3.1,ceo2浓度:30%,zeta电位:正)。
对150g该溶胶进行搅拌,向其中添加23g(摩尔比[癸酸]/[ce]=0.073)癸酸(碳数为10的直链型脂肪酸)的正丁醇溶液(浓度:20%),搅拌30分钟(相当于工序a)。
接下来,继续搅拌的同时添加4g(摩尔比[甲基丙烯酸]/[ce]=0.13)甲基丙烯酸,搅拌30分钟(相当于工序x)。停止搅拌后静置,得到透明的上清液和糊状的沉淀物。
在对1l的纯净水(去离子水)进行搅拌时滴下该沉淀物,得到粉末状沉淀。用布氏漏斗过滤该粉末状沉淀,再通入1l的纯净水进行清洗(相当于工序y)。
接下来,进行脱水、干燥,得到前体粉末(相当于工序b)。此时,进行干燥,使前体粉末的金属化合物浓度为,ceo2浓度:70%。
接下来,在该前体粉末中添加摩尔比[甲基丙烯酸]/[ce]=0.13当量的甲基丙烯酸,充分混合(相当于工序c)。其后,在80℃的干燥机内干燥1小时,得到表面改性氧化锆颗粒。
(比较例1)
将实施例1中得到的前体粉末在80℃的干燥机内干燥1小时,得到表面改性氧化锆颗粒。
(比较例2)
除代替癸酸的正丁醇溶液(浓度:20%),添加24g(摩尔比[戊酸]/[zr]=0.13)戊酸(碳数为5的直链型脂肪酸)的正丁醇溶液(浓度:20%)以外,与实施例1相同,得到表面改性氧化锆颗粒。
(比较例3)
除代替癸酸的正丁醇溶液(浓度:20%),添加25g(摩尔比[12-羟基硬脂酸]/[zr]=0.027)12-羟基硬脂酸(碳数为18的直链型脂肪酸)的正丁醇溶液(浓度:12%)以外,与实施例1相同,得到表面改性氧化锆颗粒。
(比较例4)
除在前体粉末中添加摩尔比[醋酸]/[zr]=0.15当量的冰醋酸以外,与实施例1相同,得到表面改性氧化锆颗粒。
(比较例5)
除在前体粉末中添加摩尔比[甲酸]/[zr]=0.18当量的88%甲酸以外,与实施例1相同,得到表面改性氧化锆颗粒。
(比较例6)
除代替癸酸的正丁醇溶液(浓度:20%),添加25g(摩尔比[甲基丙烯酸]/[zr]=0.13)甲基丙烯酸的正丁醇溶液(浓度:15%),搅拌30分钟以外,与实施例1相同,得到表面改性氧化锆颗粒。
(比较例7)
在按照专利文献1的实施例1所述的方法得到的氧化锆溶胶(zro2:30%,ph3.3,zeta电位:正)中添加摩尔比[柠檬酸]/[zr]=0.3当量的无水柠檬酸,接着,加入氨水调整ph为9.0。再通过超滤进行纯化及浓缩,得到氧化锆溶胶(zro2:30%,ph7.8,zeta电位:负)。
除使用150g该溶胶(zeta电位:负)作为起始原料以外,与实施例1相同,想要制备前体粉末,但羧酸与该溶胶未能充分进行反应,几乎未得到前体粉末。
<红外吸收光谱的测量>
通过以下的装置及测量条件对实施例1-实施例7中得到的表面改性金属化合物颗粒及比较例1-比较例6中得到的表面改性金属化合物颗粒的红外吸收光谱进行测量。
结果,在实施例1-7中,在1700cm-1附近观察到属于cooh的c=o拉伸振动的峰值。另外,在1560cm-1附近观察到属于coo-的不对称拉伸振动的峰值。
另一方面,在比较例1-6中,在1700cm-1附近未观察到属于cooh的c=o拉伸振动的峰值。
<红外吸收光谱测量装置及测量条件>
装置:傅里叶变换红外分光光度计,ft/ir-620(日本分光社制造)
测量条件:
方法:atr法(使用atr-one单元)
累计次数:64次
分解:4cm-1
试样:%t
背景:单
干涉仪:
灵敏度:自动
孔径:7.1mm
干涉仪速度:自动
滤光器:自动
切趾函数:余弦函数
零填充:开启
检测器:tgs
这里,实施例1的红外吸收光谱以图1中的光谱b表示,比较例1的红外吸收光谱以图1中的光谱a表示。
在实施例1的光谱b中,在1700cm-1附近观察到属于cooh的c=o拉伸振动的峰值。另外,在同一光谱中,还在1560cm-1附近观察到属于coo-的不对称拉伸振动的峰值。
另一方面,在比较例1的光谱a中,在1700cm-1附近未观察到属于cooh的c=o拉伸振动的峰值,但在1560cm-1附近观察到属于coo-的不对称拉伸振动的峰值。但是,1560cm-1的峰值的高度低于光谱a。
通过实施例1和比较例1的对比可知:在进行工序b前,不存在羧酸型的羧酸,但在进行工序b后,存在羧酸型的羧酸。另外也可知,与进行工序b前相比,进行工序b后羧酸盐型的羧酸的量增加了。
这里,工序b是添加第一羧酸的工序。如此一来,该变化仅依赖于第一羧酸的增加。其结果可知,在工序b后,添加的第一羧酸的至少一部分以羧酸型的形式存在,且剩余的第一羧酸以羧酸盐型的形式存在。
另外,用纯净水清洗实施例1提供的表面改性氧化锆颗粒,其后,以上述装置及测量条件对红外吸收光谱进行测量。该红外吸收光谱以图1中的光谱c表示。
光谱c成为了与光谱a几乎相同的光谱。由此可知,羧酸盐型的第一羧酸和羧酸型的第一羧酸这两者洗脱,成为与光谱a相同的、只有羧酸盐型的第二羧酸吸附在金属化合物颗粒表面的状态。
<粒径(中值粒径)的测量>
(实施例1-实施例6)
通过在甲苯中添加得到的表面改性氧化锆颗粒并进行搅拌,得到zro2浓度60%的溶胶。其后,在使用以下的装置及测量条件,通过动态光散射法测量该溶胶的粒径(中值粒径)时,实施例1中为9nm,实施例2中为13nm,实施例3中为10nm,实施例4中为15nm,实施例5中为10nm,实施例6中为9nm。
<粒径(中值粒径)测量装置及测量条件>
装置:粒度测量装置,zetasizernanozs(马尔文)
测量条件:
氧化物浓度:30%
材料:
zro2(氧化锆溶胶)
ri:2.17
吸收率:0.01
ceo2(二氧化铈溶胶)
ri:2.15
吸收率:0.01
分散剂:甲苯
温度:25℃
样品池:pcs1115(10mm玻璃方形样品池)
测量角度:173°
测量时间:自动
对大颗粒延长时间:no
定位方法:寻求最佳位置
自动衰减选项:yes
分析模型:通用
(实施例7)
通过在甲苯中添加得到的表面改性氧化锆颗粒并进行搅拌,得到ceo2浓度50%的溶胶。其后,在使用与上述相同的装置及测量条件,通过动态光散射法测量该溶胶的粒径(中值粒径)时,测得的粒径为9nm。
(比较例1-6)
虽然在甲苯中添加得到的表面改性氧化锆颗粒并进行了搅拌,但沉淀物剩余较多,未能分散。因此,未进行粒径的测量。
(比较例7)
如上所述,由于未得到前体粉末本身,因此也未得到表面改性氧化锆颗粒。因此,未进行粒径的测量。
<表面改性金属化合物颗粒的粘度的测量>
在以下述装置及下述测量条件,测量实施例1的表面改性金属化合物颗粒的粘度时,测得的粘度为38mpa·s。
<粘度测量装置及测量条件>
装置:vibroviscometersv-10a(a&d)
测量条件:
zro2浓度:60%
分散介质:甲苯
温度:25℃
<对表面改性金属化合物颗粒的分散性的评价(测量溶解或者分散在甲苯中时的透光率)>
在以下述装置及下述测量条件,测量实施例1的表面改性金属化合物颗粒的透光率时,在波长400nm时为25%,在波长800nm时为72%。
<透光率测量装置及测量条件>
装置:v-750(日本分光)
测量条件:
zro2浓度:60%
测量模式:%t
带宽:2nm
扫描速度:200nm/min.
响应:fast
数据间隔:0.5nm
校准:基准线
温度:20-25℃
样品池:10mm石英玻璃方形样品池
- 还没有人留言评论。精彩留言会获得点赞!