制备陶瓷微珠的方法和陶瓷微珠及其应用与流程
本发明涉及制备陶瓷微珠生坯领域,具体涉及一种制备陶瓷微珠的方法和陶瓷微珠及其应用。
背景技术:
随着材料科学的高速发展,要求高纯超细粉体的地方越来越多,对生产超细粉体的研磨介质-实心致密的陶瓷微珠的需求越来越大,很多超细材料,如涂料、油墨、染料、陶瓷粉体、电子材料、磁性材料、化妆品等都需要用研磨微珠进行超细粉碎,因此对研磨微珠的耐磨性和尺寸提出更高的要求。传统制备陶瓷球的方法有模压法、粒化-滚动法、挤压-滚动法、熔融法、溶胶-凝胶法等,然而,在制备高密度、小尺寸尤其是直径小于2毫米的陶瓷微珠方面,目前这些传统方法还难以达到。
cn1935478a公开了“一种注凝成型制备陶瓷微珠的方法及其装置”。该方法利用液滴进入加热的油相形成小球,并利用有机单体的聚合而达到固化,采用振荡装置分散小球。这种分散小球的方法对浆料的性能提出很高的要求,必须是低粘度的。一些高比表面的超细粉体配制的高固含量浆料,由于其粘度较高,用这种振荡的方法,不易分散成球。cn1468826a提供了一种采用胶态法制备陶瓷微珠的方法及装置,该方法利用液滴进入加热的油相形成小球,并利用有机单体的聚合而达到固化,采用能够调节直径的漏斗来滴出小球。这两种方法制备的陶瓷微珠在未干燥时是软、弹的小球,在冲洗等后处理过程中易造成微珠坯体表面缺陷,加大了微珠生坯的不圆度,收集的微珠表面粘附油性物质,不仅造成油性物质的浪费,还给烧结带来不便。
技术实现要素:
本发明的目的是为了克服现有技术存在的制备过程中不易分散成球,形成的微珠生坯不圆度高、硬度低,微珠生坯表面粘附油性物质,既造成油的浪费,又不利于烧结的问题,提供一种制备陶瓷微珠的方法和陶瓷微珠及其应用,以实现制备的陶瓷微珠表面光滑、干净,不圆度低,密度高,强度高,耐磨性好的目的。
为了实现上述目的,本发明一方面提供一种制作陶瓷微珠的方法,该方法包括以下步骤:
(1)配制含有有机单体、交联剂、水溶性海藻酸盐、陶瓷粉体、烧结助剂、引发剂和分散剂的浆料,所述有机单体、交联剂、水溶性海藻酸盐、引发剂和分散剂溶解在所述浆料中,其中,所述水溶性海藻酸盐的数均分子量不低于4000;
(2)将步骤(1)配制的浆料以液滴的形式送入含有催化剂的油相,进行一次注凝成型,得到固化微珠;
(3)将步骤(2)所得固化微珠送入钙源的水溶液,进行二次注凝成型,得到硬化微珠;
(4)将步骤(3)所得硬化微珠进行烧结。
本发明第二方面提供由上述方法制作的陶瓷微珠。
本发明第三方面提供上述陶瓷微珠在研磨超细粉体中的应用。
采用本发明的技术方案,制备的陶瓷微珠具有较高的致密度、强度和耐磨度,且陶瓷微珠的表面粗糙度较低,不圆度低,表面光洁;本发明的技术方案油相损耗较低,节省了成本。
附图说明
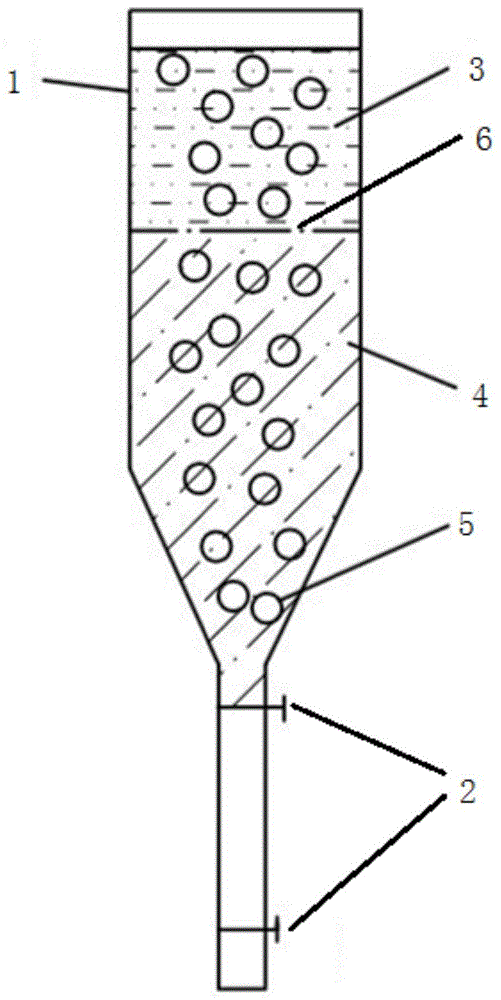
图1是本发明一种优选实施方式用于制备陶瓷微珠的设备示意图。
附图标记说明
1、塔体2、阀门3、油相
4、钙源的水溶液5、硬化微珠6、油水两相界面
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
第一方面,本发明提供了一种制作陶瓷微珠的方法,该方法包括以下步骤:
(1)配制含有有机单体、交联剂、水溶性海藻酸盐、陶瓷粉体、烧结助剂、引发剂和分散剂的浆料,所述有机单体、交联剂、水溶性海藻酸盐、引发剂和分散剂溶解在所述浆料中,其中,所述水溶性海藻酸盐的数均分子量不低于4000;
(2)将步骤(1)配制的浆料以液滴的形式送入含有催化剂的油相,进行一次注凝成型,得到固化微珠;
(3)将步骤(2)所得固化微珠送入钙源的水溶液(水相),进行二次注凝成型,得到硬化微珠;
(4)将步骤(3)所得硬化微珠进行烧结。
本发明中,为了提高所得浆料的稳定性和均匀性,优选地,步骤(1)中,配制浆料的方式包括先配制含有有机单体、交联剂、水溶性海藻酸盐和水的预混液,然后将分散剂、陶瓷粉体和烧结助剂与所述预混液进行混合,得到悬浮液,再将所述引发剂分散在所得悬浮液中得到浆料。
本发明中,优选地,先将引发剂配成浓度为10重量%的引发剂水溶液,然后将所得引发剂水溶液分散在所述悬浮液中。
本发明中,为了进一步提高所得浆料的稳定性和均匀性,优选地,步骤(1)中,配制浆料的方式还包括将分散剂、陶瓷粉体和烧结助剂与所述预混液混合后进行球磨处理。
优选地,所述球磨处理的条件包括:球磨时间为1-15min,转速为1000-2000rpm。
本发明中,优选地,步骤(1)中,配制浆料的方式还包括将所得浆料进行除泡处理。
本发明中,对所述引发剂进行分散处理的方式没有特别的限制,只要能将引发剂分散均匀即可,例如采用搅拌、涡旋或振荡的方式。
本发明中,对所得浆料进行除泡处理的方式没有特别的限制,只要能将浆料中气泡除去即可,例如可以选择消泡剂除泡和/或真空搅拌除泡。
本发明中,对所述真空搅拌除泡没有特别的限制,可以为本领域常规使用的真空搅拌除泡方法,比如在压力为0.001-0.005mpa、转速为50-100rpm下除泡1-15min。
优选地,所述消泡剂选自磷酸三丁酯、正辛醇和甲基硅油中的1-2种。
本发明中,优选地,有机单体和交联剂的总重量含量、水溶性海藻酸盐的重量含量、引发剂的重量含量之间的比值为18-350:0.2-10:1,更优选为50-250:0.6-7:1,更优选地,所述有机单体和交联剂的总重量含量、水溶性海藻酸盐的重量含量、引发剂的重量含量之间的比值为(50、70、90、110、120、145、170、190、210、230、250):(0.6、0.8、1、1.5、1.6、1.7、1.8、1.9、3、5、7):1。
优选地,所述陶瓷粉体、有机单体与交联剂的重量比为400-6000:20-60:1,更优选地,所述陶瓷粉体、有机单体与交联剂的重量比为(400、800、1200、1600、2000、2500、3000、3500、4000、4500、5000、5500、6000):(20、25、30、35、40、45、50、55、60):1。
优选地,相对于100重量份的陶瓷粉体,烧结助剂的用量为0.05-5重量份。
优选地,相对于100重量份的陶瓷粉体,分散剂的用量为0.05-2重量份。
优选地,所述陶瓷粉体的体积占陶瓷粉体的体积与预混液的体积之和的40-60%,优选为45-55%。
本发明中,陶瓷粉体的体积通过陶瓷粉体的质量与陶瓷粉体的密度的比值所得到,所述预混液的体积为在常温常压条件下,通过预混液的质量与预混液的密度的比值所得到。
本发明中,对所述水溶性海藻酸盐的数均分子量没有特别的限制,为了提高所述硬化微珠的强度,优选地,所述水溶性海藻酸盐的数均分子量为4000-185000,更优选为30000-150000。
本发明中,优选地,所述水溶性海藻酸盐选自海藻酸钠和/或海藻酸钾。
本发明中,优选地,所述有机单体选自具有自由基活性的有机单体,更优选地,所述具有自由基活性的有机单体选自丙烯酰胺、丙烯酸羟丙酯、甲基丙烯酰胺、甲基丙烯酸羟乙酯、甲基丙烯酸羟丙酯和n-羧甲基丙烯酰胺中的至少一种。
本发明中,对所述交联剂没有特别的限制,可以为本领域常规使用的交联剂,为了提高交联效果,优选地,所述交联剂选自n,n-亚甲基双丙烯酰胺,本发明中,优选地,所述烧结助剂选自cao、mgo、sio2、tio2、mno2和稀土氧化物中的至少一种,更优选地,所述cao由碳酸钙、石灰和硅灰石中的至少一种提供,mgo由碱式碳酸镁、烧滑石、菱镁矿和轻质氧化镁中的至少一种提供,sio2由硅微粉、硅灰石和烧滑石中的至少一种提供,稀土氧化物优选选自y2o3、la2o3和ceo2中的至少一种。
本发明中,所述引发剂可以为过硫酸盐,优选地,所述引发剂选自过硫酸钾、过硫酸钠和过硫酸铵中的至少一种,更优选地,所述引发剂选自过硫酸铵。
优选地,所述分散剂选自聚丙烯酸钠、聚丙烯酸铵、聚丙烯酸、柠檬酸铵、羧甲基纤维素和聚丙烯酰胺中的至少一种。
优选地,所述陶瓷粉体选自碳化硅(sic)、α-氧化铝(α-al2o3)、γ-氧化铝(γ-al2o3)、二氧化锆(zro2(3y))、氮化硅(si3n4)、硅酸锆(zrsio4)、氮化铝(aln)和钛酸铝(al2tio5)中的至少一种。
本发明中,为了提高注凝成型的效果,优选地,所述陶瓷粉体的平均粒径为0.3-45μm,优选为0.5-15μm,纯度不小于99重量%,优选为99.5-99.95重量%。
本发明中,为了进一步提高注凝成型的效果,优选地,步骤(2)中所述液滴的直径为0.4-3mm,优选为0.6-2mm。
本发明中,对步骤(2)中所述一次注凝成型的条件没有特别的限制,为了提高注凝成型的效果,优选地,步骤(2)中所述一次注凝成型的条件包括:所述液滴在油相中的停留时间为30-120s,优选为40-110s;所述油相的温度为40-90℃,优选为45-80℃。
本发明中,对步骤(3)中所述二次注凝成型的条件没有特别的限制,为了提高注凝成型的效果,优选地,步骤(3)中所述二次注凝成型的条件包括:所述固化微珠在水相中停留时间为5-20s,优选为8-15s;所述水相的温度为35-65℃,优选为38-55℃。
本发明中,为了提高一次注凝成型的效果,优选地,所述油相的温度比水相的温度高5-25℃。
本发明中,为了提高一次注凝成型的效果,优选地,所述油相中的油选自二甲基硅油、乙基硅油或石蜡油,更优选选自二甲基硅油。
本发明中,为了进一步提高一次注凝成型的效果,优选地,所述油相中的油选自粘度为300-1000mpa·s的二甲基硅油。
本发明中,对所述催化剂的来源没有特别的限制,为了提高催化效果,优选地,所述催化剂选自n,n,n,n-四甲基乙二胺。
本发明中,为了进一步提高催化效果,优选地,相对于100重量份的油相,所述油的用量为97.5-99.6重量份,催化剂的用量为0.4-2.5重量份。
本发明中,对所述钙源没有特别的限制,为了提高二次注凝效果,优选地,所述钙源选自水溶性钙盐,更优选自氯化钙、硝酸钙、碳酸氢钙、硫酸氢钙、亚硫酸氢钙、溴化钙和碘化钙中的至少一种。
本发明中,为了进一步提高二次注凝效果,优选地,所述钙盐水溶液中钙离子的含量为0.2-20重量%。
本发明中,对所述烧结的条件没有特别的限制,为了提高烧结的效果,优选地,所述烧结的条件包括:烧结温度为1400-2000℃,烧结时间为0.5-4h。
本发明中,为了进一步提高注凝成型陶瓷微珠的效果,优选地,所述注凝成型陶瓷微珠的过程还可以在如图1所示的设备中进行,该设备包括塔体1、阀门2以及塔体1内部的如上所述的油相3和钙源的水溶液4;
其中,所述油相3底部界面与钙源的水溶液4顶部界面接触形成油水两相截面6;
所述油相3用于将进入的浆料液滴进行一次注凝成型,得到固化微珠;
所述钙源的水溶液4用于将所得固化微珠进行二次注凝成型,得到硬化微珠5。
本发明中,对所述油相3的深度和钙源的水溶液4的深度没有特别的限制,为了进一步提高注凝成型陶瓷微珠的效果,优选地,所述油相3的深度使得所述浆料液滴在油相3中的停留时间为30-120s,优选为40-110s;所述钙源的水溶液4的深度使得所述固化微珠在钙源的水溶液4中的停留时间为5-20s,优选为8-15s。
本发明第二方面提供了一种由上述方法制作的陶瓷微珠。
本发明第三方面提供了一种上述陶瓷微珠在研磨超细粉体中的应用。
以下将通过实施例对本发明进行详细描述。以下实施例中,
不圆度参数通过圆度仪直接测得;
抗压强度参数参考国标《铁矿球团抗压强度测定方法》(gb/t14201-93)测得;
耐磨度以磨损率表示,磨损率参照建材行业标准《耐磨氧化铝球》(jc/t848.1-2010)测得,转速为100rpm,填充密度为2.2kg/l,
磨损率=(m1-m2)÷(m1×t),
m1,m2:材料磨损前后的质量,单位为g,
t:研磨时间,单位为h;
油相损耗指的是每制备1千克陶瓷微珠,油相的减少量;
硅酸锆粉为山东盛太锆业资源有限公司牌号为sg-01的市售品;
碳化硅粉为山东金蒙新材料有限公司公司牌号为w0.5的市售品;
菱镁矿为泽镁科技公司牌号为ar的市售品,氧化镁含量为40-44.5重量%;
la2o3为上海邦景实业有限公司公司牌号为br的市售品,纯度为99重量%;
zro2(3y)为焦作李封工业有限责任公司公司牌号为lf-3y-2#的市售品;
海藻酸钠为天津市光复精细化工研究所公司牌号为cp的市售品;
二甲基硅油为广州穗欣化工有限公司牌号为201的市售品,粘度为200-1000mpa·s;
圆度仪购自广州威而信精密仪器有限公司牌号为dtp-2000c的市售品;
球磨机购自佛山市业津机电设备有限公司牌号为yjks-2000的市售品。
实施例1
(1)配制100g预混液,其中各组分的重量含量为:丙烯酰胺10%,n,n-亚甲基双丙烯酰胺0.4%,海藻酸钠(数均分子量为100000)0.12%,余量为水;
将315.4g的α-氧化铝(α-al2o3,平均粒径为1μm,纯度为99.95%)、1.9g的菱镁矿(氧化镁含量为42重量%)和1.6g的柠檬酸铵加入预混液中,在球磨机中,在2000rpm下,快速球磨5min,得到悬浮液;其中,α-氧化铝的体积占α-氧化铝的体积与预混液的体积之和的45%;
然后将质量浓度为10%的过硫酸铵水溶液加入悬浮液中,搅拌均匀,然后在0.001mpa、转速70rpm下真空除泡10min,得到浆料,过硫酸铵加入量为浆料总质量的0.017%。
(2)将上述浆料加到滴定器中,使分散成直径1.3mm的小液滴,滴落到成型塔中的油相中(含有99重量%的二甲基硅油(粘度为500mpa·s)和1重量%的催化剂n,n,n,n-四甲基乙二胺)进行一次注凝成型,得到固化微珠,液滴在二甲基硅油中停留时间为60s,二甲基硅油温度为50℃。
(3)所得固化微珠进入质量浓度为5%的cacl2的水溶液中,进行二次注凝成型,得到硬化微珠;固化微珠在水溶液中的停留时间为10s,水溶液温度保持在40℃。
(4)所得硬化微珠经阀门采出后依次进行清洗和干燥,然后在1600℃条件下,烧结2小时,得到陶瓷微珠。
所得陶瓷微珠的指标见表1。
实施例2
(1)配制100g预混液,其中各组分的重量含量为:丙烯酰胺12%,n,n-亚甲基双丙烯酰胺0.25%,海藻酸钠(数均分子量为50000)0.16%,余量为水;
将323.6g的α-氧化铝(平均粒径为1μm,纯度为99.95重量%)、76g的二氧化锆(zro2(3y),平均粒径为0.8μm,纯度为99.5重量%)、0.4g的la2o3和4g的聚丙烯酸铵加入预混液中,在球磨机中,在1500rpm下,快速球磨15min,得到悬浮液;其中,α-氧化铝和二氧化锆(zro2(3y)的总体积占α-氧化铝的体积、二氧化锆(zro2(3y)的体积与预混液的体积之和的50%;
然后将质量浓度为10%的过硫酸铵溶液加入悬浮液中,搅拌均匀,然后加入消泡剂甲基硅油搅拌,除泡,得到浆料,过硫酸铵加入量为浆料总质量的0.02%。
(2)将上述浆料加到滴定器中,使分散成直径0.8mm的液滴,将液滴滴落到成型塔中的油相中(含有99.5重量%的二甲基硅油(粘度为300mpa·s)和0.5重量%的催化剂n,n,n,n-四甲基乙二胺)进行一次注凝成型,得到固化微珠,液滴在二甲基硅油中停留时间为45s,二甲基硅油温度为70℃。
(3)所得固化微珠进入质量浓度为7%的ca(no3)2的水溶液中,进行二次注凝成型,得到硬化微珠;固化微珠在水溶液中的停留时间为10s,水溶液温度保持在45℃。
(4)所得硬化微珠经阀门采出后依次进行清洗和干燥,然后在1550℃条件下,烧结1.5小时,得到陶瓷微珠。
所得陶瓷微珠的指标见表1。
实施例3
(1)配制100g预混液,其中各组分的重量含量为:丙烯酰胺15%,n,n-亚甲基双丙烯酰胺0.6%,海藻酸钠0.22%,余量为水;
将456的硅酸锆(平均粒径为0.9μm,纯度为99.5%)和5g的聚丙烯酸钠加入预混液中,在球磨机中,在1500rpm下,快速球磨3min,得到悬浮液;其中,硅酸锆的体积占硅酸锆的体积与预混液的体积之和的50%;
将质量浓度为10%的过硫酸铵溶液加入悬浮液中,然后加入消泡剂正辛醇,搅拌均匀,然后在0.003mpa、转速50rpm下真空除泡5min,得到浆料,过硫酸铵加入量为浆料总质量的0.026%。
(2)将上述浆料加到滴定器中,使分散成直径1.6mm的液滴,将液滴滴落到成型塔中的油相中(含有98.8重量%的二甲基硅油(粘度为500mpa·s)和1.2重量%的催化剂n,n,n,n-四甲基乙二胺)进行一次注凝成型,得到固化微珠,液滴在二甲基硅油中停留时间为100s,二甲基硅油温度为65℃。
(3)所得固化微珠进入质量浓度为3%的ca(clo3)2的水溶液中,进行二次注凝成型,得到硬化微珠;固化微珠在水溶液中的停留时间为5s,水溶液温度保持在50℃。
(4)所得硬化微珠经阀门采出后依次进行清洗和干燥,然后在1500℃条件下,烧结4小时,得到陶瓷微珠。
所得陶瓷微珠的指标见表1。
实施例4
(1)配制100g预混液,其中各组分的重量含量为:丙烯酰胺13%,n,n-亚甲基双丙烯酰胺0.4%,海藻酸钠0.28%,余量为水;
将372g的碳化硅(平均粒径为0.5μm,纯度为99.5%),4g的y2o3和5.4g(碳化硅用量的1.2%)的聚丙烯酰胺加入预混液中,在球磨机中,在转速1000rmp/h条件下,快速球磨9min,得到悬浮液;其中,碳化硅的体积占碳化硅的体积与预混液的体积之和的54%;
然后将质量浓度为10%过硫酸铵溶液加入悬浮液中,加入消泡剂磷酸三丁酯,搅拌均匀,然后在0.003mpa、转速70rpm下真空除泡8min,得到浆料,过硫酸铵加入量为浆料总质量的0.031%。
(2)将上述浆料加到滴定器中,使分散成直径2mm的液滴,将液滴滴落到成型塔中的油相中(含有98.4重量%的二甲基硅油(粘度为800mpa·s)、1.6重量%的催化剂n,n,n,n-四甲基乙二胺)进行一次注凝成型,得到固化微珠,液滴在二甲基硅油中停留时间为80s,二甲基硅油温度为55℃。
(3)所得固化微珠进入质量浓度为10%的ca(hso3)2的水溶液中,进行二次注凝成型,得到硬化微珠,固化微珠在水溶液中的停留时间为11s,水溶液温度保持在48℃。
(4)所得硬化微珠经阀门采出后依次进行清洗和干燥后在2000℃条件下,烧结2小时,得到陶瓷微珠。
所得陶瓷微珠的指标见表1。
实施例5
按照实施例1的方法制备陶瓷微珠,不同的是,浆料中海藻酸钠的含量为0.1重量%。
所得陶瓷微珠的指标见表1。
实施例6
按照实施例1的方法制备陶瓷微珠,不同的是,步骤(1)中将262.4g的α-氧化铝(平均粒径为1μm,纯度为99.95%)、1.6g的菱镁矿(氧化镁含量为42重量%)和1.6g的柠檬酸铵加入预混液中,其中,α-氧化铝的体积占α-氧化铝的体积与预混液的体积之和的40%;
所得陶瓷微珠的指标见表1。
实施例7
按照实施例1的方法制备陶瓷微珠,不同的是,步骤(2)中停留时间为30s。
所得陶瓷微珠的指标见表1。
实施例8
按照实施例1的方法制备陶瓷微珠,不同的是,油相和水相的温度均为50℃。所得陶瓷微珠的指标见表1。
对比例1
按照实施例1的方法制备陶瓷微珠,不同的是,将步骤(3)中的cacl2水溶液替换为等体积的油相(含有99重量%的二甲基硅油(粘度为500mpa·s)和1重量%的催化剂n,n,n,n-四甲基乙二胺),将步骤(2)得到的固化微珠直接进行步骤(4)的操作。
所得陶瓷微珠的指标见表1。
对比例2
按照实施例1的方法制备陶瓷微珠,不同的是,将海藻酸钠替换为等量的低酯果胶。
所得陶瓷微珠的指标见表1。
对比例3
按照实施例1的方法制备陶瓷微珠,不同的是,海藻酸钠的数均分子量为3500。
所得陶瓷微珠的指标见表1。
对比例4
按照实施例1的方法制备陶瓷微珠,不同的是,将浆料中的海藻酸钠替换为等量的cacl2,将步骤(3)中cacl2的水溶液替换为等量等浓度的海藻酸钠溶液。
所得陶瓷微珠的指标见表1。
表1
通过表1的结果可以看出,采用本发明的方法制备的陶瓷微珠的品质优于对比例的陶瓷微珠,且使用本发明的方法所用的油相的损耗量更少。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!