一种高比表面碳包裹的过渡金属碳化物材料的制备方法
本发明涉及一种高比表面碳包裹的过渡金属碳化物m(nb、mo、w)@c的制备方法,涉及新型材料的合成。
背景技术:
过渡金属碳化物(tmc)是由金属元素以及碳元素形成的一系列具有类金属性质的间隙化合物,由于其结构中通常存在共价键网络,材料的共价键作用很强,因此tmc具有很高的硬度、机械强度以及良好的催化活性(氢解反应、逆水煤气反应、电催化反应等)。
通常,金属碳化物是将金属氧化物与碳在1800-2000oc的惰性气体中进行高温渗碳制得。但该制备过程能耗高,而且高的合成温度容易导致所得金属碳化物颗粒团聚,比表面积低。cn111203249a专利公开了“一种石墨烯包覆过渡金属碳化物纳米胶囊的制备方法及其在微波催化领域的应用”,将过渡金属块放在直流电弧氢等离子体设备上,通入氢气、惰性气体和含碳元素气体使腔体总气压达0.005-9.5*104pa,接通电源形成稳定电弧,蒸发过渡金属块,得到石墨烯包覆过渡金属碳化物纳米胶囊,该方法虽然可以得到过渡金属碳化物,但制备过程中使用甲烷,乙烷等易燃气体,其次使用直流电弧氢等离子体设备等,生产成本较高。cn101371988a专利公开了“一种过渡金属碳化物催化材料的制备方法及应用”,该方法以炭材料、氧化物或分子筛等为载体,以过渡金属化合物为前驱体,通过浸渍或机械混合载体和过渡金属前躯体,采用微波辅助的化学沉积方法制备负载型过渡金属碳化物催化剂,材料比表面积为69m2/g。cn109201002a专利公开了“一种炭包裹的过渡金属碳化物复合材料、制备方法及其吸附用途”,该复合材料是以tialc2、葡萄糖、纤维素或木屑为原料,碱性溶液为溶剂,在120-220oc,24-96小时条件下水热/溶剂热法一步制备获得。此体系中,碱性溶液naoh作为刻蚀剂将tialc2中的al溶解,最终形成棒状和纤维状的炭包裹复合材料(记为:c@naoticx)。
由此可见,目前过渡金属碳化物材料的合成存在能耗高,过程复杂(需要碱液刻蚀),比表面积小等问题。针对这些现有技术的不足,我们通过采用合适的有机物作为碳源,水溶性金属盐作为前驱体,将两者混合水热,并对水热产物进行碳热还原反应得到碳包裹的过渡金属碳化物。与已报道的金属碳化物合成方法相比,该方法操作简单,能耗较低,可制备具有高比表面积的过渡金属碳化物材料。
技术实现要素:
基于上述目的,本发明提供了一种能耗较低的高比表面碳包裹的过渡金属碳化物m(nb,mo,w)@c材料的制备方法。采用水溶性金属盐作为前驱体,糖或醛与酚作为碳源,水热晶化后在惰性气氛下碳热还原反应制得该材料。其中本过渡金属碳化物材料合成过程具有操作简单,能耗较低,金属碳化物材料比表面积高的优点。
本发明包括过渡金属碳化物催化剂的合成方法,其核心是在相对温和的条件下,以糖或醛与酚有机聚合物作为碳源,碳源与金属前驱相互作用,在碳热过程中,金属前驱形成的金属氧化物还原并与碳源中的炭发生反应,最终合成一种具有高比表面积的、碳包裹的过渡金属碳化物m(nb,mo,w)@c材料,合成该金属碳化物材料的方法未见报道。
本发明的优势在于:
(1)原料易得。采用糖类和酚类等由可再生木质生物质制得的有机物作为碳源,绿色、廉价可再生;
(2)采用水热法,制备工艺简单,过程中不使用强酸强碱试剂,环境友好;
(3)能耗较低。大多数过渡金属碳化物的合成要经过1500-2000℃的高温焙烧,本发明焙烧温度范围在800-1200℃,能耗降低。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
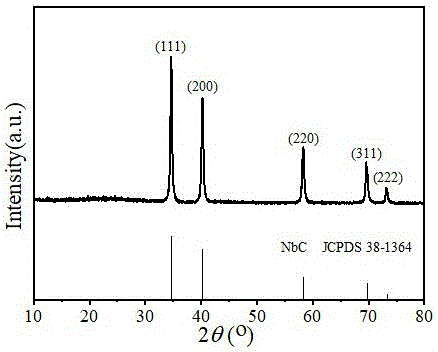
图1为本发明实施例1的碳包覆碳化铌纳米颗粒nbc@c-1的x射线衍射图。
图2为本发明实施例11中800℃碳化制备的nbc@c-11的x射线衍射图。
图3为本发明实施例12的碳包覆碳化钼纳米颗粒mo2c@c-1的x射线衍射图。
图4为本发明实施例15的碳包覆碳化钨纳米颗粒wc@c-1的x射线衍射图。
图5为本发明实施例1的nbc@c-1材料扫描电镜图。
图6为本发明实施例1的nbc@c-1材料透射电镜图。
图7为本发明实施例12的moc@c-1材料透射电镜图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚明白,以下结合具体实施例,并参照附图,对本发明进一步详细说明。
上述材料的一个典型制备方法如下:
(1)称量金属盐前驱体与醛/糖类、酚类有机物混合,加入100ml去离子水。金属前驱体与有机物总重的质量比值在0.05-0.5之间;
(2)将混合物溶液于50℃下水浴搅拌0.5小时;
(3)将步骤(2)处理得到的悬浮液转移到聚四氟乙烯水热釜中,140-200℃条件下,反应12到72小时;
(4)通过抽滤回收固体沉淀物,在60℃条件下干燥24小时;
(5)将(4)中获得的固体产物研磨后,在800-1200℃,惰性气氛中碳热还原反应0.5-12小时。
实施例1
将0.4g草酸铌(v)粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为nbc@c-1。对材料进行表征测量以确定其特征。得到的x射线粉末衍射图见图1,经x射线粉末衍射鉴定为碳化铌。扫描电镜图见图5。透射电镜图见图6。
实施例2
将0.4g五氯化铌粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为nbc@c-2。经x射线粉末衍射鉴定为碳化铌。
实施例3
将0.4g草酸铌铵粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化12小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为nbc@c-3。经x射线粉末衍射鉴定为碳化铌。
实施例4
将0.4g草酸铌(v)粉末,2g甲醛与2g苯酚与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为nbc@c-4。经x射线粉末衍射鉴定为碳化铌。
实施例5
将0.4g草酸铌(v)粉末,2g木糖与2g对苯二酚与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化78小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为nbc@c-5。经x射线粉末衍射鉴定为碳化铌。
实施例6
将0.2g草酸铌(v)粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为nbc@c-6。经x射线粉末衍射鉴定为碳化铌。
实施例7
将2g草酸铌(v)粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为nbc@c-7。经x射线粉末衍射鉴定为碳化铌。
实施例8
将0.4g草酸铌(v)粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。140℃水热晶化24小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为nbc@c-8。经x射线粉末衍射鉴定为碳化铌。
实施例9
将0.4g草酸铌(v)粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。200℃水热晶化24小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为nbc@c-9。经x射线粉末衍射鉴定为碳化铌。
实施例10
将0.4g草酸铌(v)粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1200℃。煅烧时间为4小时。样品标记为nbc@c-10。经x射线粉末衍射鉴定为碳化铌。
实施例11
将0.4g草酸铌(v)粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤的方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到铌-有机聚合物前体。将铌-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至800℃。煅烧时间为12小时。样品标记为nbc@c-11。经x射线粉末衍射鉴定为五氧化二铌。
实施例12
将0.6g钼酸铵粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将4g葡萄糖与4g间苯二酚粉末与上述溶液混合,继续水热搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤等方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到钼-有机聚合物前体。将钼-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为12小时。样品标记为mo2c@c-1。对材料进行表征测量以确定其特征。得到的x射线粉末衍射图见图3,经x射线粉末衍射鉴定为mo2c。透射电镜图见图7。
实施例13
将0.6g六羰基钼(mo(co)6)粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将4g葡萄糖与4g间苯二酚粉末与上述溶液混合,继续水热搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤等方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到钼-有机聚合物前体。将钼-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为mo2c@c-2。经x射线粉末衍射鉴定为mo2c。
实施例14
将0.6g仲钼酸铵粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将4g葡萄糖与4g间苯二酚粉末与上述溶液混合,继续水热搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤等方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到钼-有机聚合物前体。将钼-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为6小时。样品标记为mo2c@c-3。经x射线粉末衍射鉴定为mo2c。
实施例15
将0.6g偏钨酸铵粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤等方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到钨-有机聚合物前体。将钨-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为12小时。样品标记为wc@c-1。对材料进行表征测量以确定其特征。得到的x射线粉末衍射图见图4,经x射线粉末衍射鉴定为wc/w2c混合相。
实施例16
将0.6g钨酸铵粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤等方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到钨-有机聚合物前体。将钨-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为12小时。样品标记为wc@c-2。经x射线粉末衍射鉴定为wc/w2c混合相。
实施例17
将0.6g六羰基钨(w(co)6)粉末,2g葡萄糖与2g间苯二酚粉末与100ml去离子水混合,将混合物溶液于50℃下水浴搅拌1小时。将混合液转移到聚四氟乙烯内衬中,置于不锈钢钢晶化釜内。160℃水热晶化24小时。通过离心抽滤等方式回收固体沉淀物,用去离子水不断洗涤。最后将产品在60℃鼓风干燥箱干燥24小时,得到钨-有机聚合物前体。将钨-聚合物前体粉碎成粉末,置于刚玉瓷舟中,在惰气气氛下,管式炉焙烧至1000℃。煅烧时间为12小时。样品标记为wc@c-3。经x射线粉末衍射鉴定为wc/w2c混合相。
表1为本发明实施例1、实施例12和实施例15的nbc@c-1、moc@c-1和wc@c-1的bet结果。
表1nbc@c材料的比表面积。
由表1可知,实施例1的材料nbc@c-1比表面积约为513m²/g,实施例12的材料moc@c-1比表面积约为463m²/g,实施例15的材料wc@c-1比表面积约为525m²/g。
图1为实施例1的碳包覆碳化铌纳米颗粒nbc@c-1的x射线衍射图,由图中观察到五组衍射峰((111),(200),(220),(311),(222)),确定碳包覆碳化铌材料结构中主相为nbc。
图2为实施例11中,800℃碳热还原得到的材料nbc@c-11的x射线衍射图,由图中观察到材料的主晶相为五氧化二铌,未生成nbc相。
图3为实施例12的碳包覆碳化钼纳米颗粒mo2c@c-1的x射线衍射图,由图中观察到八组衍射峰((100),(002),(101),(102),(110),(103),(112),(201)),确定碳包覆碳化钼材料结构中主相为mo2c。
图4为实施例15的碳包覆碳化钨纳米颗粒wc@c-1的x射线衍射图,由图中观察到的衍射峰((001),(100),(101),(110),(002),(111),(200),(102)),确定碳包覆碳化钨材料结构中含有wc相;由图中观察到的衍射峰((100),(002),(101),(102),(110),(103),(200),(112),(201))确定碳包覆碳化钨材料结构中含有w2c相。
图5为实施例1的nbc@c-1材料扫描电镜图,图中可看出材料表面较为均匀,呈现球状的结构。
图6为实施例1的nbc@c-1材料透射电镜图,可以看出碳化铌纳米颗粒为球状,均匀分布在材料表面,颗粒尺寸大概在10-20nm之间,外面包覆了碳膜。
图7为实施例12的moc@c-1材料透射电镜图,可以看出碳化钼纳米颗粒为球状,均匀分布在材料表面,颗粒尺寸大概在10-15nm之间,外面包覆了碳膜。
- 还没有人留言评论。精彩留言会获得点赞!