一种碳氮包覆的Fe0.4Co0.6S2@NC中空纳米盒子的制备方法及其应用
本发明属于纳米材料制备技术领域,特别涉及一种碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子材料的制备方法及其应用。
背景技术:
普鲁士蓝(pb)及其类似物(pba)因其本身的开放框架结构、理论比容量高、安全性且易于合成而成为有前途的钠存储材料。pb/pba的一般通式为axma[mb(cn)6]y·zh2o,其中a是嵌入在骨架间隙位置的碱金属阳离子;ma和mb表示通过氰基配体连接的过渡金属,分别与氰基基团中的氮和碳八面体配位,ma和mb可以是相同或不同的元素。然而,pb/pba经常面临导电性差、结构不稳定性等困扰。为解决这些问题,人们已投入大量精力来优化pb/pba的晶体结构和导电性,例如将其与高导电性碳材料复合、合成空心结构等。普鲁士蓝衍生出的各种金属纳米结构,如金属硫化物、金属硼化物和碳包覆的金属/金属合金等,可以提供高表面积,更高效地应用于电化学领域。
技术实现要素:
本发明的目的是基于以现有手段合成的pb/pba比表面积低、导电性差等问题,提出了一种以普鲁士蓝为牺牲模板、过程简单可控的碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子的制备方法。
本发明提供一种碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子材料的制备方法,包括以下步骤:
a.将氯化钴和柠檬酸三钠在去离子水中通过搅拌得到清澈均一的溶液a,将铁氰化钠超声溶于去离子水中得到均一溶液b,b加入a中,搅拌后静置得到前驱体钴基普鲁士蓝材料;
b.将步骤a中得到的前驱体溶解于去离子水,加入多巴胺水溶液,并加入氨基丁三醇调节ph,搅拌使前驱体表面包覆多巴胺,冷干得到粉末样品;
c.将步骤b所得包覆多巴胺后的普鲁士蓝前驱体与过量硫粉混合,在惰性气体保护下退火,即可获得碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子。
所述步骤a中使用的柠檬酸三钠、氯化钴和铁氰化钠均为化学纯度。
在一个优选的实验方案中,步骤a中所用柠檬酸三钠、氯化钴和铁氰化钠的摩尔比为10:(1~2):1,优选为10:2:1。
优选的,步骤a中a和b溶液去离子水的体积均为200~300ml,优选为250ml,搅拌转速均为400~500rpm,转速过大,容易导致溶液溅出。
优选的,步骤a中静置时间为24~48h。
所述步骤b中使用的多巴胺和氨基丁三醇均为化学纯度;
优选的,步骤b中前驱体和多巴胺的质量比范围为0.5~4,前驱体水溶液体积为100~300ml,优选为200ml,多巴胺水溶液体积为20~50ml,优选为30ml。
优选的,步骤b中多巴胺水溶液通过超声方法得到,所述超声时间为20~40min。
优选的,步骤b中氨基丁三醇浓度为10mm,搅拌转速为400~500rpm,转速过大,容易导致溶液溅出。
优选的,步骤c中硫粉与包覆多巴胺后的普鲁士蓝前驱体质量比≥2:1。
优选的,步骤c中升温前需提前预通氩气20~30min,排除石英管中的氧气。
优选的,步骤c中退火温度为400~600℃,退火时间为3h。
本发明还提供了一种电池负极的制备方法,其特征在于,先根据上文所述制备方法得到碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子活性材料,然后再将其制成电池负极。
本发明还提供一种如上所述方法制备碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子的应用,先按照以上所述的制备方法制备得到碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子活性材料,然后再将所述材料制成电池负极。
本发明公开了一种碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子的制备方法。其工艺流程为:a)按摩尔比称取柠檬酸三钠、氯化钴和铁氰化钠,搅拌使溶于去离子水中,避光静置,得到钴基普鲁士蓝前驱体;b)通过搅拌的方法将前驱体表面均匀包覆多巴胺;c)将包覆后的普鲁士蓝与硫粉混合,通过高温煅烧得到碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子。所述前驱体和多巴胺的质量比为1:(0.25~2);所述退火的温度为400~600℃。本发明提供的制备方法简单可行,过程可控,可批量制备,得到的fe0.4co0.6s2@nc中空纳米盒子在可充电电池、电容器和电化学催化等领域具有巨大的应用前景,尤其具有优异的电化学性能。
附图说明
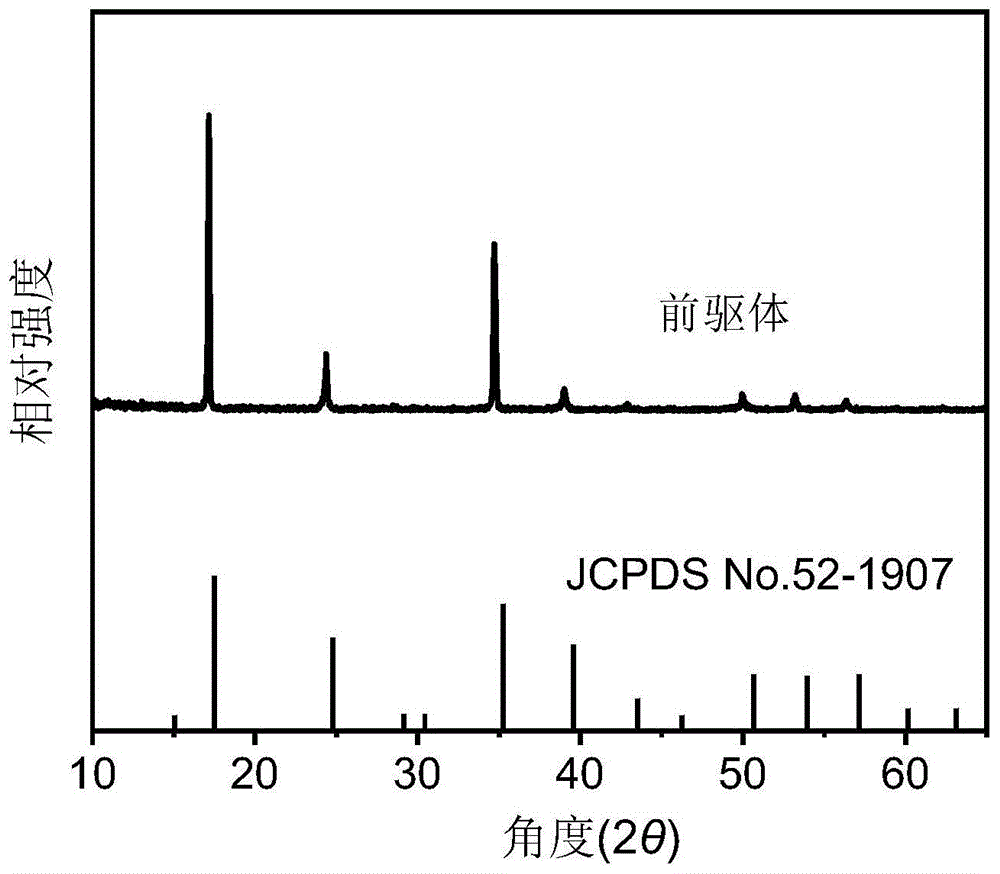
图1为本发明的普鲁士蓝前驱体的x射线衍射图谱;
图2为本发明的普鲁士蓝前驱体的扫描电子显微镜图;
图3为本发明实施例1得到的fe0.4co0.6s2@nc中空纳米盒子的x射线衍射图谱;
图4为本发明实施例1得到的fe0.4co0.6s2@nc中空纳米盒子的扫描电子显微镜图;
图5为本发明实施例1得到的fe0.4co0.6s2@nc中空纳米盒子的透射电子显微镜图;
图6为本发明实施例1得到的fe0.4co0.6s2@nc中空纳米盒子的temmapping图;
图7为本发明实施例2得到的fe0.4co0.6s2@nc中空纳米盒子的x射线衍射图谱;
图8为本发明实施例2得到的fe0.4co0.6s2@nc中空纳米盒子的透射电子显微镜图;
图9为本发明实施例3得到的fe0.4co0.6s2@nc中空纳米盒子的x射线衍射图谱;
图10为本发明得到的fe0.4co0.6s2@nc中空纳米盒子钠离子电池性能数据图。
具体实施方式
下面结合附图及具体实施例详细介绍本发明。但以下的实施例仅限于解释本发明,本发明的保护范围应包括权利要求的全部内容,而且通过以下实施例的叙述,本领域的技术人员是可以完全实现本发明权利要求的全部内容。
本发明提供一种碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子材料的制备方法,包括以下步骤:
a.将氯化钴和柠檬酸三钠在去离子水中通过搅拌得到清澈均一的溶液a,将铁氰化钠超声溶于去离子水中得到均一溶液b,b加入a中,搅拌后静置得到前驱体钴基普鲁士蓝材料;
b.将步骤a中得到的前驱体溶解于去离子水,加入多巴胺水溶液,并加入氨基丁三醇调节ph,搅拌使前驱体表面包覆多巴胺,冷干得到粉末样品;
c.将步骤b所得包覆多巴胺后的普鲁士蓝前驱体与过量硫粉混合,在惰性气体保护下退火,即可获得碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子。
所述前驱体和多巴胺的质量比为1:(0.25~2);所述退火的温度为400~600℃。
本发明中,多巴胺水溶液通过超声方法得到,所述超声的频率优选为50~100hz,更优选为60~90hz;所述超声的时间优选为2~4小时。若不超声或超声时间不够,粉末不能够充分溶解,会影响最终样品中碳氮包覆层的厚度,从而影响性能。
本发明中,所述磁力搅拌的转速优选为400~500rpm。
本发明将所述前驱体和最终样品进行干燥获得,所述干燥的温度优选为50~100℃,更优选为80~100℃;所述干燥的时间优选为12~24小时,更优选为15~20小时。本发明优选将所述前驱体和最终样品置于真空烘箱中进行烘干。
本发明中,所述前驱体和多巴胺的质量比优选为1:(0.25~2),更优选为1:1。
本发明中,所述硫粉与包覆多巴胺后的普鲁士蓝前驱体质量比≥2:1,保证硫源的充足。
本发明中,所述退火的温度优选为400~600℃,更优选为450~550℃,最优选为500℃;所述温度过高,所烧样品结块严重,温度过低,结晶性不好;所述退火的时间优选为3小时。
本发明还提供了一种电池负极的制备方法,本发明中的电池负极中包含活性材料,所述活性材料为上文所述制备方法制备得到的碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子材料,先根据上文所述制备方法得到碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子活性材料,然后再将其制成电池负极。
本发明对于碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子制备成电池负极的具体方法没有特殊的限制,采用本领域常用的电池负极材料和制备方法即可。
与现有技术相比,本发明的优点是:
1.本发明生产工艺流程简单,产量可控。
2.本发明中使用的原料来源广泛、价格便宜,所以生产成本低。
3.本发明生产过程安全,在惰性气体保护下退火,安全无害。
4.本发明在实验过程中对环境无害,不产生废液、废料。
5.本发明生产的fe0.4co0.6s2@nc中空立方纳米盒子结构增加pb/pba的比表面积,硫化增加导电性;前驱体普鲁士蓝中钴元素还可被其他过渡金属取代,增加可调节性;用途广泛,不仅可以用于储能领域的电极材料,还可以用于催化、复合材料等领域。
为了进一步说明本发明,以下结合实施例对本发明提供进一步详细描述,但不以任何方式限制本发明的范围。
下面实施例中所用的设备及测试方法如下:
(1)x射线衍射仪(d8-advance,
(2)扫描电子显微镜(15kv,jeol,jsm-6700f);
(3)透射电子显微镜(jeol,jem2010);
(4)land电池测试仪(ct2001a,武汉市蓝电电子股份有限公司)。
实施例1
a.称取3.675g(12.5mmol)柠檬酸三钠(c6h5na3o7·2h2o)和594.8mg(2.5mmol)氯化钴(cocl2·6h2o)加入250ml去离子水中,500rpm转速下搅拌至溶解,得到均一溶液a;称取605.08mg(1.25mmol)铁氰化钠(na4[fe(cn)6]·10h2o)加入250ml去离子水中,500rpm转速下搅拌至溶解,得到均一溶液b;将b倒入a中,继续搅拌约5min至液体浑浊,避光静置24h。
b.待上述混合物料产生沉淀后,舍弃上层清液,用去离子水和无水乙醇对沉淀产物清洗数次,放入80℃烘箱中干燥12h,得到前驱体钴基普鲁士蓝纳米立方块。
c.称取200mg上述前驱体粉末,搅拌溶于200ml去离子水中,并加入约90mg氨基丁三醇(c4h11no3);另称取200mg多巴胺超声溶于30ml去离子水中;在持续搅拌(500rpm)下,将多巴胺溶液逐滴加入前驱体溶液中,继续搅拌4h至混合溶液完全变黑,用去离子水洗涤数次,冷冻后放入冷干机冷干。
d.称取160mg上述多巴胺包覆的前驱体粉末和350mg硫粉,放入磁舟中,氩气气氛下500℃退火3h;用二硫化碳和无水乙醇洗涤数次,目的是去除过量的硫;洗涤后放入80℃烘箱中干燥12h,得到碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子。
图1和图2分别为前驱体的x射线衍射谱及扫描电镜图,从图中可以证明成功合成了前驱体钴基普鲁士蓝纳米立方块。
图3为所制备的碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子的x射线衍射谱,说明样品中同时含有cos2和fes2。
图4和图5分别为所制备的碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子的扫描电子显微镜图和透射电子显微镜图,说明合成了边长约500nm的中空立方纳米盒子。
图6为所制备的碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子的temmapping图,各元素的均匀分布证明了fe0.4co0.6s2@nc中空纳米盒子的制备成功。
电极片制作:将fe0.4co0.6s2@nc中空纳米盒子、导电剂科琴黑、粘结剂pvdf以7:2:1的质量比在nmp(1-甲基-2吡咯烷酮)中充分混合研磨得到均一的浆料,并将该浆料涂抹在集流体铜网上,然后在70℃烘箱中预干燥30分钟,最后在100℃的真空烘箱中完全干燥24h,即可制得电极片;
电池组装:将上述所得fe0.4co0.6s2@nc电极片为负极、钠片为正极、1m高氯酸钠为电解液、whatman玻璃纤维膜为隔膜在手套箱中组装成2032型纽扣电池。
电池测试:将上述所制得的纽扣电池在武汉蓝电系统进行测试,室温恒定在25℃,性能测试数据如图10所示。
实施例2
按照实施例1中的方法制备得到碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子,不同的是,本实例中步骤c中前驱体和多巴胺的质量分别为200mg和100mg。
图7和图8分别为本发明实施例2得到的碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子样品的x射线衍射图谱和透射电子显微镜图,由图7可知,前驱体和多巴胺质量比为1:0.5时,制得的样品中仍然同时含有cos2和fes2,但从图8可以看出,由于碳包覆量少,中空立方结构不能得到很好保持。
实施例3
按照实施例1中的方法制备得到碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子,不同的是,本实例中步骤d中退火温度为400℃。
图9为本发明实施例3得到的碳氮包覆的fe0.4co0.6s2@nc中空纳米盒子样品的x射线衍射图谱,由图可知,退火温度为400℃、时间3h时,制得的样品中仍然同时含有cos2和fes2,但由于温度较低,反应未完全,30°附近含有杂相。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!