一种碳酸盐前驱体及其制备方法与流程
本发明属于锂离子电池材料技术领域,具体涉及一种碳酸盐前驱体及其制备方法。
背景技术:
发展新能源汽车是我国由汽车大国迈向汽车强国的必由之路。然而,续航里程短和成本居高不下制约了新能源汽车的快速发展。随着国家补贴政策对新能源车续航里程的要求,行业洗牌加剧,开发高性能动力电池已成为企业在激烈竞争中生存下来的唯一途径。目前全球普遍使用的动力电池正极材料由最初的锰酸锂、磷酸铁锂逐步过渡到镍钴锰酸锂、镍钴铝酸锂等三元材料,能量密度在逐渐升高,但安全性却有待继续改善。富锂锰基材料作为新一代的锂电正极材料,使用了大量的锰元素,与licoo2和三元材料相比,不仅价格低,而且安全性好、对环境友好,能够很好满足电动汽车、储能电站和小型电子产品等领域的使用要求,是未来最具有发展前景的动力电池正极材料之一。然而,富锂锰基材料目前仍然有不尽如人意之处,首次效率、倍率性能、循环稳定性以及循环过程中的电压逐渐衰减、阻抗增加等,很大程度上限制了富锂锰基正极材料的商业化和工业化应用。
富锂锰基材料可以采用多种方法制备,但是具有工业意义的合成方法仅限于采用共沉淀法合成前驱体再结合混锂高温烧结法制备。通过共沉淀法制备的前驱体根据沉淀剂的不同又可以分为氢氧化物体系和碳酸盐体系。按照当前三元前驱体材料的氢氧化物共沉淀工艺,制备的富锂锰基材料电化学性能并不理想,主要原因是mn容易被氧化而导致前驱体分相,烧结产物很容易形成li2mno3团簇。在共沉淀过程中采用n2气体保护并且调整络合剂摩尔比可以解决这个问题,但生产综合成本升高。另外,在采用氢氧化物共沉淀体系中,反应体系ph高、腐蚀性强,且大量使用氨水作络合剂,虽然能制备致密程度高、形貌优选的前驱体,但含氨废水的处理需要增加除氨设备,增加了生产成本。所以,期望开发一种低成本的无氨沉淀法。
就目前而言,电化学性能好的富锂锰基材料一般都是采用碳酸盐共沉淀工艺制备前驱体,也是产业化研究最多的技术路线。但是碳酸盐共沉淀法也存在工艺不稳定、颗粒形貌难调控、振实密度低、并且杂质含量较高的问题,所以富锂锰基正极材料前驱体的生产工艺仍然需要深入研究,产品批次的一致性问题仍然需要改善。
公开号为cn108557905a的中国专利申请公开了一种富锂锰基材料前驱体及其制备方法、富锂锰基正极材料及其制备方法、锂电池,通过碳酸盐共沉淀法控制混合金属离子浓度、沉淀剂浓度、络合剂浓度、混合速率、搅拌速率、反应ph和反应温度,生成叶片状的富锂锰基材料碳酸盐前驱体;该专利提供的前驱体的一次纤维堆叠较疏松,有利于叶片状形貌的产生,致密程度较低,前驱体的振实密度较低,使用该前驱体经混锂煅烧后,最终制备出单晶富锂锰基正极材料。公开号为cn106797016a的中国专利申请公开了锂镍锰钴氧化物阴极材料的碳酸盐前驱物及其制造方法,采用连续碳酸盐共沉淀的无氨体系,通过引入晶种的方式,制备了具有较宽分布的碳酸盐前驱体,该前驱体随着晶种加入量的不同,d50不同,对应粒度分布也不同。公开号为cn106795008a的中国专利申请公开了具有优选形态的含杂质阴极材料及自含杂质金属碳酸盐的制备方法,通过引入晶种,并控制摩尔流速比co3/m,制备了碳酸盐前驱体,所述碳酸盐前驱体中,钠硫摩尔比0.4<na/s<2,0.4weight%<2*na+s<1.6weight%,前驱体中钠、硫含量较高,且实施例中尽管前驱体粒度分布较宽,但振实密度仍偏低。
技术实现要素:
针对现有技术中制备的碳酸盐前驱体致密度较低、杂质含量高、生产成本高等问题,本发明提供一种碳酸盐前驱体及其制备方法。本发明所述的制备方法工艺简单、生产成本低廉,且能制备得到稳定性好、性能优异的球形碳酸盐前驱体。
为解决上述技术问题,本发明提供以下技术方案。
首先,本发明提供一种碳酸盐前驱体,其化学通式为mn1-x-ycoxniyatco3,式中,0<x<0.5,0<y<0.5,0<t<0.05。该碳酸盐前驱体的一次颗粒为纤维状,一次颗粒从中心向外部呈辐射堆积成纺锤体珊瑚状二次球形颗粒。该碳酸盐前驱体的粒度为3~25μm,比表面积为10~55m2/g,振实密度为1.1~2.0g/cm3。该碳酸盐前驱体含有na及s杂质,这些杂质的含量及比例达到非常低的水平,均在1000ppm以下。基于某些良好的工艺设计,杂质含量甚至可以达到更低。
基于同样的发明构思,本发明另提供上述的富锂锰基碳酸盐前驱体的制备方法,该方法采用碳酸盐共沉淀结晶,共沉淀反应以可溶性碳酸盐为沉淀剂,通过低温成核和高温生长并使用提浓设备延长颗粒的生长,生成致密程度高、振实密度大的中间体,中间体再经水热反应得到杂质含量低的球形碳酸盐前驱体材料。
具体来说,本发明所述的碳酸盐前驱体的制备方法包括以下步骤:
s1,配制包括ni、co、mn和m的混合盐溶液,a选自mg、b、al、cr、zr、ti、w、v中的一种以上;
配制包含碳酸根离子的沉淀剂溶液;
配制包含f离子的络合剂溶液;
s2,共沉淀反应,包括成核和生长两个阶段;
成核阶段:将步骤s1配制的混合盐溶液、沉淀剂溶液和络合剂溶液同时加入到反应釜中,控制反应温度为25-55℃,反应ph值为10.0-11.0,搅拌速率为300-800rpm/min,待反应浆料粒度达到目标值,成核反应结束;
生长反应阶段:将步骤s1配制的混合盐溶液、沉淀剂溶液和络合剂溶液同时加入到反应釜中,控制反应温度为35-65℃,反应ph值为9.5-10.5,搅拌速率为200-500rpm/min,待反应浆料粒度达到目标值,将反应浆料固液分离,所得固相经陈化洗涤后,加水调成浆料,并转移至水热反应釜中,在150-250℃下反应一定时间,随后自然冷却至室温;
s3,将步骤s2经水热反应后的物料固液分离,所得固相经干燥后即为所述的碳酸盐前驱体。
上述的制备方法中,进一步地,步骤s1中,配制的混合盐溶液中的总的金属离子浓度为0.5-4mol/l,沉淀剂溶液中碳酸根离子的浓度为0.5-4mol/l,络合剂溶液中f离子的浓度为0.1-1mol/l。
进一步的,混合盐溶液中的总的金属离子浓度为1-3mol/l,更优选为2-3mol/l;沉淀剂溶液中碳酸根离子的浓度优选为1-3mol/l,更优选为2-3mol/l;络合剂溶液中f离子的浓度优选为0.1-0.5mol/l,更优选为0.1-0.3mol/l。
进一步的,配制混合盐溶液时,镍盐选自硫酸镍、氯化镍、硝酸镍及乙酸镍中的一种或者几种;钴盐选自硫酸钴、氯化钴、硝酸钴及乙酸钴中的一种或者几种;锰盐选自硫酸锰、氯化锰、硝酸锰及乙酸锰中的一种或几种。
配制混合盐溶液使用的可溶性镍盐、钴盐、锰盐的阴离子优选为相同的阴离子,在共沉淀反应过程中,采用相同的阴离子盐可以减少杂质的引入,使共沉淀反应后母液中留下的可溶物更单一,减少沉淀物的分离,同时便于可溶物的回收,降低生产成本及有利于环境保护。
进一步的,配制沉淀剂时,沉淀剂选自碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾中的一种或几种。
进一步的,配制络合剂时,络合剂选自氟化钠、氟化钾、氟化铵中的一种或几种。
在上述的制备方法中,进一步的,所述反应釜为连续搅拌反应釜。
在上述的制备方法中,进一步的,在步骤s2的生长阶段,控制反应体系的液固的质量比为1-19:1。
在上述的制备方法中,进一步的,在步骤s2的生长阶段,通过提浓设备控制反应体系的液固比,反应釜溢流出的浆料通过提浓设备提浓、固液分离后的固相返回到生长反应阶段,循环往复,直至达到目标粒径。
本发明在共沉淀反应中,采用可溶性碳酸盐为沉淀剂,成核反应阶段和生长反应阶段独立进行,在不同阶段通过调节和控制反应的关键参数,比如搅拌速度、ph和反应温度等,并通过提浓设备将反应生产的母液及时排出,调节料浆中的固体含量等,通过将反应结束后的沉淀物分离、洗涤后调制成一定浓度的浆液,转移至水热反应釜进行水热反应后,分离、洗涤烘干后即可得到纤维状形貌的从中心向外部辐射堆积而成的球形富锂锰基碳酸盐前驱体。
与现有技术相比,本发明所述的制备方法工艺简单、成本低廉,且制备得到的富锂锰基碳酸盐前驱体杂质含量低。
附图说明
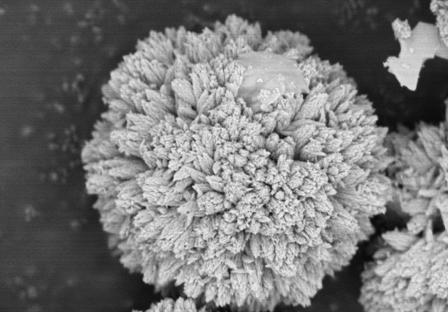
图1为实施例1制备得到的碳酸盐前驱体的sem图。
图2为实施例1制备得到的碳酸盐前驱体的xrd图。
具体实施方式
下面结合附图对本发明进行详细描述,本部分的描述仅是示范性和解释性,不应对本发明的保护范围有任何的限制作用。此外,本领域技术人员根据本文件的描述,可以对本文件实施例中以及不同实施例中的特征进行相应组合。
碳酸盐前驱体从原理上可以通过将mso4及na2co3和络合剂按不同流速连续泵入连续搅拌反应釜中进行沉淀反应来获得;但在实际情况中,钠和硫总是被包裹在颗粒内,或者嵌入到晶体结构中,可能的反应方程式为:
(1-x)mso4+(1-y)na2co3=(m1-xna2x)[(co3)1-y(so4)y]+(1-x-y)na2so4,式中,x、y<0.01;杂质含量对正极材料的性能有显著影响,杂质含量需要控制在较低水平才能获得性能优异的正极材料。
一般而言,碳酸盐前驱体中na/s=2x/y可表征杂质的存在形式。本发明的碳酸盐前驱体中na/s控制在0.5~10范围内。当na/s在0.5~2时,说明硫酸钠被包裹在颗粒内部或吸附在颗粒表面;当na/s>2时,说明不但有硫酸钠被包裹在颗粒内部,同时还有钠嵌入到晶格中。本发明提供的技术方案,na和s的含量满足公式:2*na+s<0.5weight%。
获得的碳酸盐前驱体的比表面积通过标准bet方法在3h-2000beta型比表面测试系统上进行测量;粒径在malvernmastersizer2000上进行测试;成分及杂质在pe-8000的icp-aes系统上进行测试;振实密度在百特bt-301上进行测试。
本发明通过蠕动泵将混合金属离子溶液、沉淀剂溶液、络合剂溶液同时泵入到连续搅拌反应釜中,控制沉淀剂溶液和混合金属离子溶液的流速比,保证反应体系的ph值,先后分阶段进行成核反应和生长反应,同时,通过与反应釜相连通的浓密设备将反应釜中产生的母液除去,使总进料流量和母液排出流量相等以达到浓密设备中的液位保持不变,产生的物料全部被截留在合成釜内,整体的料浆浓度会逐渐升高,且颗粒也在不断生长直至颗粒长大到目标值;接着将反应釜中的物料分离、洗涤除杂后,加水调浆泵入水热反应釜中,维持一定的温度和时间进行水热反应;随后,将反应后的物料分离、洗涤烘干后即可得到低杂质含量的纤维状一次颗粒堆积成呈纺锤体珊瑚状二次球形颗粒的碳酸盐前驱体。
在本发明提供的碳酸盐前驱体制备方法中,反应釜中物料的停留时间由粒度生长至目标值时的整体的料浆浓度和成核反应阶段结束时的晶核粒度和数量决定。假定每小时向反应釜中提供的物料的质量为m,成核反应结束时的料浆浓度为m1、粒径为d1,生长到目标粒度时的料浆浓度为m2,粒径为d2,则停留时间t=100*(m2-m1)/m(h)。因此可以通过改变泵速即每小时提供的质量或者流速及晶核数量来改变停留时间。成核反应阶段的沉淀剂溶液和混合金属离子溶液的流速比远大于1,温度控制在25~55℃,ph控制在10.00~11.00;生长反应阶段沉淀剂溶液和混合金属离子溶液的流速比大于1,优选大于1且低于1.5,温度控制35~65℃,ph控制在9.50~10.50。在成核反应阶段,泵入到反应釜中的混合盐溶液和沉淀剂的量的比值控制在远大于1,优选地,控制在>3以上。随着反应的进行,反应釜中的ph会随着反应的进行缓慢降低。随后,调整沉淀剂和混合金属溶液的流量比、温度及ph等参数进行生长反应,优选co3/m大于1,特别优选控制在0.9~1.2范围内。沉淀反应结束后,将物料分离、洗涤并调浆后泵入水热反应釜中,在150℃~250℃、优选在180℃~220℃温度下,反应2~6h,自然冷却至室温后洗涤烘干即可得到杂质钠硫含量低的纤维状一次颗粒堆积成呈纺锤体珊瑚状二次球形颗粒的碳酸盐前驱体。
为了更好的解释本发明,以便于理解,下面通过具体的实施方式对本发明作详细的描述。
实施例1
①进料溶液的配制:将硫酸锰、硫酸钴、硫酸镍、硫酸镁按化学计量摩尔比mn:co:ni:a=0.70:0.15:0.15:0.01溶解在去离子水中配制成总金属离子浓度为2mol/l的混合金属离子溶液。将碳酸钠溶解在去离子水中配制成2mol/l的沉淀剂溶液。将氟化钠溶解在去离子水中配制成0.2mol/l的络合剂溶液。
②共沉淀反应:成核反应阶段,将碳酸钠溶液、混合金属离子溶液按碳酸根离子与总的金属离子的摩尔比=3:1调节泵入碳酸钠溶液和混合金属离子溶液的流速,络合剂在总进料溶液中的浓度控制在0.01mol/l,同时泵入到100l连续搅拌反应釜中,反应釜中盛有50l碳酸钠溶液作为底液;控制成核反应温度30℃,搅拌转速600rpm,进料一段时间后,随着沉淀反应的进行,ph缓慢降低,并在11.00~10.50之间变化,通过粒度分析仪实时监测反应釜中的料浆粒度,直至粒度图中的粒度分布呈正态分布,成核反应阶段完成,此时粒度<2微米。粒度生长反应阶段,调整上一阶段的碳酸根离子和总的金属离子的摩尔比为1.3:1、搅拌转速为500rpm及反应温度45℃,随着沉淀反应的进行,ph缓慢降低,并在10.50~9.80之间变化。由于反应过程中,反应釜的溢流口关闭,通过浓密设备(沉降槽)将母液从反应釜中连续排出,反应料浆被截留在反应釜内无物料排出,直至长到目标粒度。最终物料的粒度分布由成核反应阶段的粒度分布决定。
③生长反应结束后的料浆经分离、陈化洗涤后加水调浆并转移至水热反应釜中,控制水热反应温度分别为150℃、180℃和200℃,水热反应时间2h。
对比例1
对比例1与实施例1相比,没有步骤③。
检测分析实施例1和对比例1制备得到的碳酸盐前驱体的td、d50、bet、na和s含量,结果如表1所示。
表1碳酸盐前驱体的物化指标
通过实施例1和对比例1制备得到的碳酸盐的物化指标对比,可发现,水热反应对于降低碳酸盐前驱体中的na的含量至关重要。
图1是实施例1采用水热反应温度为180℃时制备得到的碳酸盐前驱体的sem图,从图中可以看出,碳酸盐前驱体的二次颗粒为纺锤体珊瑚状,由纤维状一次颗粒辐射状态堆积而成。
图2是实施例1采用水热反应温度为180℃时制备得到的碳酸盐前驱体的xrd图,从图中可以看出制备得到的前驱体是纯相,且结晶度高。
实施例2:
①进料溶液的配制:将硫酸锰、硫酸钴、硫酸镍、硫酸铝按化学计量比mn:co:ni:a=0.60:0.20:0.20:0.01溶解在去离子水中配制成总金属离子浓度为2mol/l的混合金属离子溶液。将碳酸钠溶解在去离子水中配制成2mol/l的沉淀剂溶液。将氟化钠溶解在去离子水中配制成0.2mol/l的络合剂溶液。
②共沉淀反应:成核反应阶段,将碳酸钠溶液、混合金属离子溶液按碳酸根离子和总的金属离子的摩尔比为5:1调节物料的泵入流速,络合剂在总进料溶液中的浓度控制在0.012mol/l,同时泵入到100l连续搅拌反应釜中,反应釜中盛有50l碳酸钠溶液作为底液;控制成核反应温度40℃,搅拌转速800rpm,进料一段时间后,随着沉淀反应的进行,ph缓慢降低,并在11.00~10.80之间变化,通过粒度分析仪实时监测反应釜中的料浆粒度,直至粒度图中的粒度分布呈正态分布,成核反应阶段完成,此时粒度<2微米。粒度生长反应阶段,调整上一阶段的碳酸根离子和总的金属离子的摩尔比为1.2:1、搅拌转速为600rpm及反应温度55℃,随着沉淀反应的进行,ph缓慢降低,并在10.80~10.20之间变化。由于反应过程中,反应釜的溢流口关闭,通过浓密设备(沉降槽)将母液从反应釜中连续排出,反应料浆被截留在反应釜内无物料排出,直至长到目标粒度。
③生长反应结束后的料浆经分离、陈化洗涤后加水调浆并转移至水热反应釜中,控制水热反应温度分别为180℃、200℃和220℃,水热反应时间2h。
对比例2-1:
对比例2-1与实施例2相比,区别在于没有步骤③的过程。
对比例2-2:
对比例2-2的步骤③的水热反应温度为200℃,与实施例2相比,区别在于:在生长反应阶段,打开溢流口,未使用浓密设备(沉降槽)将料浆截留在反应釜内,颗粒的停留时间急速降低,且生长较快。
对比例2-3:
对比例2-3的步骤③的水热反应温度为200℃,与实施例2相比,区别在于:在生长反应阶段,未向反应釜中泵入氟化钠络合剂,在沉淀反应过程中,反应釜中游离的锰离子事先未被氟离子络合,而是和钴离子、镍离子共同同碳酸根离子结合形成沉淀,生长较快。
检测分析将实施例2和对比例2制备得到的碳酸盐前驱体的td、d50、bet、na和s含量,结果如表2所示。
表2碳酸盐的物化指标
通过实施例2和对比例2-1、2-2和2-3的对比,可以明显得出,本发明提供的技术方案对于碳酸盐前驱体的振实密度的提高、以及na和s含量的降低,具有特别重要的意义。
实施例3:
①进料溶液的配制:将硫酸锰、硫酸钴、硫酸镍、硫酸镁按化学计量比mn:co:ni:a=0.50:0.20:0.30:0.01溶解在去离子水中配制成总金属离子浓度为2mol/l的混合金属离子溶液。将碳酸钠溶解在去离子水中配制成2mol/l的沉淀剂溶液。将氟化钠溶解在去离子水中配制成0.2mol/l的络合剂溶液。
②共沉淀反应:成核反应阶段,将碳酸钠溶液、混合金属离子溶液按碳酸根离子和总的金属离子的摩尔比为3:1调节物料的泵入流速,络合剂在总进料溶液中的浓度控制在0.01mol/l,同时泵入到100l连续搅拌反应釜中,反应釜中盛有50l碳酸钠溶液作为底液;控制成核反应温度45℃,搅拌转速700rpm,进料一段时间后,随着沉淀反应的进行,ph缓慢降低,并在10.80~10.00之间变化,通过粒度分析仪实时监测反应釜中的料浆粒度,直至粒度图中的粒度分布呈正态分布,成核反应阶段完成,此时粒度<2微米。粒度生长反应阶段,调整上一阶段的碳酸根离子和总的金属离子的摩尔比为1.1:1、搅拌转速为600rpm及反应温度65℃,随着沉淀反应的进行,ph缓慢降低,并在10.00~9.50之间变化。由于反应过程中,反应釜的溢流口关闭,通过浓密设备(沉降槽)将母液从反应釜中连续排出,反应料浆被截留在反应釜内无物料排出,直至长到目标粒度。最终物料的粒度分布由成核反应阶段的粒度分布决定。
③生长反应结束后的料浆经分离、陈化洗涤后加水调浆并转移至水热反应釜中,控制水热反应温度分别为150℃、180℃和200℃,水热反应时间2h。
对比例3:
对比例3与实施例3的区别在于:没有步骤③,直接得到碳酸盐前驱体。
检测分析实施例3和对比例3制备得到的碳酸盐前驱体的td、d50、bet、na和s的含量,结果如下表3所示。
表3碳酸盐物化指标
从表3的物化指标数据,进一步说明水热反应对碳酸盐前驱体中na含量降低的重要性。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!