一种丝光沸石分子筛及制备方法和应用
1.本技术涉及一种丝光沸石分子筛及制备方法和应用,属于沸石分子筛材料技术领域。
背景技术:
2.丝光沸石分子筛是一类重要的硅铝沸石分子筛材料,作为重要的吸附和催化材料广泛应用于石油加工和精细化工领域。丝光沸石分子筛骨架结构归属于正交晶系,cmcm空间群。其骨架由沿c轴方向平行的12元环和8元环孔道组成,两者通过沿b轴方向的8 元环侧口袋相连接。事实上,由于沿c轴方向的8元环孔道太窄导致无法穿透大多数分子,丝光沸石分子筛在实际的催化反应中多表现为一维孔道沸石分子筛的特点。丝光沸石分子筛独特的孔道结构和酸性质,使得其广泛应用于甲苯歧化制二甲苯,甲醇和氨制甲胺,二甲醚羰基化制乙酸甲酯等催化反应过程。
3.丝光沸石已被证明是二甲醚羰基化反应的有效分子筛催化剂。特别是丝光沸石显示出高的二甲醚羰基化活性和乙酸甲酯选择性。先前的工作已经证明,二甲醚在酸性沸石上的羰基化机理涉及二甲醚在b酸位上的吸附形成甲氧基,并随后与co反应生成乙酰基中间体,进而与二甲醚反应生成乙酸甲酯。其中,乙酰基的生成是整个反应的决速步骤。此外,研究发现羰基化的活性中心位于8元环侧口袋内,而12元环主孔道的酸性位会引起副反应,导致催化剂积碳失活。由此可见,制备8元环孔道b酸比例高的丝光沸石不仅有利于提高催化剂二甲醚羰基化反应性能,还可以减少副反应的发生,提高反应的稳定性。而现有合成技术一般难以实现对铝分布的调控。因此,制备8元环孔道酸量比例高的丝光沸石分子筛具有重要意义。
技术实现要素:
4.根据本技术的一个方面,提供一种丝光沸石分子筛,所述丝光沸石分子筛以n,n,n-三甲基-1-金刚烷基氢氧化铵作为模板剂,制备得到的催化剂8元环孔道b酸比例高,具有更好的催化性能。
5.一种丝光沸石分子筛,所述丝光沸石分子筛无水化学组成表示为:
6.nr
·
mm
·
(si
x
al)o
2x+2
ꢀꢀ
式ⅰ;
7.其中,r表示模板剂,所述模板剂为n,n,n-三甲基-1-金刚烷基氢氧化铵;
8.n表示每摩尔(si
x
al)o
2x+2
中模板剂r的摩尔数,n的取值范围为 0.05≤n≤0.85;
9.m表示碱金属离子;
10.m表示每摩尔(si
x
al)o
2x+2
中碱金属离子m的摩尔数,m的取值范围为0.15≤m≤0.95;
11.x表示丝光沸石分子筛骨架中si元素和al元素的摩尔比,x的取值范围为10≤x≤30。
12.可选地,所述x的取值范围为10≤x≤24。
13.可选地,所述丝光沸石分子筛具有由圆形纳米柱聚集成团的形貌结构。
14.可选地,所述圆形纳米柱的直径约200~300nm。
15.根据本技术的一个方面,提供上述任一项所述的丝光沸石分子筛的制备方法,其特征在于,所述制备方法包括以下步骤:
16.将含有硅源、铝源、m源、晶种、水和模板剂的初始凝胶混合物晶化,得到所述丝光沸石分子筛。
17.可选地,所述硅源包括硅溶胶、白炭黑、活性二氧化硅、正硅酸酯、水玻璃、偏高岭土、高岭土中的至少一种。
18.可选地,所述铝源包括偏铝酸钠、烷氧基铝、铝盐、偏高岭土、高岭土中的至少一种。
19.可选地,所述m源为m的氢氧化物。
20.可选地,所述m的氢氧化物包括氢氧化锂、氢氧化钠、氢氧化钾、氢氧化铯中的至少一种。
21.可选地,所述晶种包括丝光沸石原粉或将丝光沸石原粉处理后得到的丝光沸石中的至少一种。
22.所述处理包括焙烧、球磨处理、碱处理、氟离子刻蚀处理中的至少一种。
23.可选地,所述初始凝胶混合物中,各组分的摩尔比为:
24.sio2/al2o3=20-60;
25.m2o/al2o3=1-20,其中m为碱金属离子;
26.r/al2o3=1-25;
27.h2o/al2o3=130-2000;
28.晶种质量/投料sio2固体质量=0.1~7%。
29.可选地,所述初始凝胶混合物中,各组分的摩尔比为:
30.sio2/al2o3=20-48;
31.m2o/al2o3=4-12;
32.r/al2o3=4-16;
33.h2o/al2o3=150-1600;
34.晶种质量/投料sio2固体质量=0.5-7%。
35.可选地,sio2/al2o3的摩尔比的下限选自20、28、36、45中的任一值; sio2/al2o3的摩尔比的上限选自28、36、45、60中的任一值。
36.可选地,m2o/al2o3的摩尔比的下限选自1.1、2.1、2.3、2.4、2.8、3、 3.2、3.6、4、4.4、4.5、5.5、6、7、7.8、8.5、10、12、15、18中的任一值;m2o/al2o3的摩尔比的上限选自2.1、2.3、2.4、2.8、3、3.2、3.6、4、 4.4、4.5、5.5、6、7、7.8、8.5、10、12、20中的任一值。
37.可选地,r/al2o3的摩尔比的下限选自1.2、1.6、2.4、3.5、4、4.3、 4.6、4.9、5.3、5.8、6.2、6.7、7.2、7.6、7.9、8.3、8.8、11.5、13、14.5、 16.0、17.5、19、20.5、22、23.5中的任一值。
38.可选地,r/al2o3的摩尔比的上限选自1.6、2.4、3.5、4、4.3、4.6、4.9、5.3、5.8、6.2、6.7、7.2、7.6、7.9、8.3、8.8、11.5、13、14.5、16.、 17.5、19、20.5、22、23.5、24中的任一值。
39.可选地,h2o/al2o3的摩尔比的下限选自140、180、224、250、390、450、490、620、700、
730、750、760、810、850、880、900、920、936、 1200中的任一值。
40.可选地,h2o/al2o3的摩尔比的上限选自180、224、250、390、450、 490、620、700、730、750、760、810、850、880、900、920、936、1200、 1950中的任一值。
41.可选地,晶种质量/投料sio2固体质量的百分比下限选自0.5wt%、 1wt%、2wt%、3wt%、4wt%、5wt%、6wt%中的任一值;晶种质量/投料 sio2固体质量的百分比的上限选自1wt%、2wt%、3wt%、4wt%、5wt%、 6wt%、7wt%中的任一值。
42.可选地,所述初始凝胶混合物通过以下步骤得到:
43.先将铝源、m源与去离子水混合,之后依次加入硅源、晶种、模板剂,搅拌,得到所述初始凝胶混合物;
44.可选地,所述晶化的条件包括:
45.晶化温度为120-220℃;
46.晶化时间为0.5-168小时。
47.可选地,所述晶化的条件包括:
48.晶化温度为140-180℃;
49.晶化时间为0.5-168小时。
50.可选地,所述晶化为动态晶化或者静态晶化。
51.可选地,晶化温度的下限独立地选自120℃、130℃、135℃、145℃、150℃、165℃、175℃、180℃、190℃、200℃中的任一值;晶化温度的上限独立地选自130℃、135℃、145℃、150℃、165℃、175℃、180℃、 190℃、200℃、220℃中的任一值。
52.可选地,晶化时间的下限独立地选自0.5h、16h、24h、32h、41h、 45h、49h、52h、58h、66h、70h、78h、90h、105h、110h中的任一值;晶化时间的上限独立地选自16h、24h、32h、41h、45h、49h、52h、 58h、66h、70h、78h、90h、105h、110h、168h中的任一值。
53.根据本技术的另一个方面,提供一种催化剂,所述催化剂由丝光沸石分子筛经离子交换、焙烧得到;
54.所述丝光沸石分子筛包括上述任一项所述的丝光沸石分子筛、根据上述任一项所述的制备方法得到的丝光沸石分子筛中的至少一种。
55.可选地,所述离子交换为铵离子交换。
56.所述焙烧的温度为400~700℃。
57.可选地,所述焙烧的温度上限选自400℃、450℃、500℃、500℃、550℃、 600℃、650℃或700℃,下限选自400℃、450℃、500℃、500℃、550℃、 600℃或650℃。
58.可选地,所述催化剂的8元环孔道中的b酸中心数量在总b酸中心数量中的占比为80%~92%。
59.本技术获得一种以n,n,n-三甲基-1-金刚烷基氢氧化铵为模板剂合成的丝光沸石分子筛,骨架中si元素和al元素的摩尔比在10-30。以此制备得到的催化剂具有良好的催化剂性能。
60.本技术制备的催化剂是具有高8元环孔道b酸比例的纯相丝光沸石分子筛晶体,分子筛8元环孔道中的b酸中心数量在丝光沸石分子筛总b 酸中心数量中的占比为80%~92%。
61.本技术制备的催化剂在二甲醚羰基化制乙酸甲酯催化反应中表现出优异的催化
性能,二甲醚的转化率可达到92%以上,乙酸甲酯选择性可达到99%以上。
62.根据本技术的另一个方面,提供一种二甲醚羰基化制乙酸甲酯的方法,所述方法包括以下步骤:
63.将含有二甲醚和一氧化碳的混合气与催化剂接触、反应,得到乙酸甲酯;
64.所述催化剂包括上述任一项所述的催化剂中的至少一种。
65.可选地,所述混合气中,二甲醚和一氧化碳的体积比为1:5~9。
66.可选地,所述反应的条件包括:
67.混合气的空速为500~7500ml g-1
h-1
。
68.反应温度为150~300℃。
69.反应压力为0.5~5mpa。
70.可选地,在与混合气接触之前,先用含吡啶的气流对所述催化剂进行处理。
71.可选地,混合气的空速选自500ml g-1
h-1
、1000ml g-1
h-1
、2000ml g-1 h-1
、3000ml g-1
h-1
、4000ml g-1
h-1
、5000ml g-1
h-1
、6000ml g-1
h-1
、7500mlg-1
h-1
中的任一值,或任意两值之间的范围值。
72.可选地,所述反应温度为150℃、200℃、250℃或300℃中的任一值,或任意两值之间的范围值。
73.可选地,所述反应压力为0.5mpa、2mpa、4mpa、5mpa中的任一值,或任意两值之间的范围值。
74.作为一种实施方案,本技术提供了一种丝光沸石分子筛,该丝光沸石分子筛为纯相的丝光沸石分子筛(骨架中si元素和al元素的摩尔比在 10-30),以n,n,n-三甲基-1-金刚烷基氢氧化铵为模板剂,具有优异的二甲醚羰基化反应性能。
75.所述丝光沸石分子筛无水化学组成表示为:
76.nr
·
mm
·
(si
x
al)o
2x+2
ꢀꢀ
式ⅰ77.其中,在式ⅰ中,r表示模板剂,所述模板剂为n,n,n-三甲基-1-金刚烷基氢氧化铵;
78.n表示每摩尔(si
x
al)o
2x+2
中模板剂r的摩尔数,n的取值范围 0.05≤n≤0.85;
79.m表示碱金属离子;
80.m表示每摩尔(si
x
al)o
2x+2
中碱金属离子的摩尔数,m的取值范围 0.15≤m≤0.95;
81.x为丝光沸石分子筛骨架中si元素和al元素的摩尔比,x的取值范围 10≤x≤30。
82.本技术可以得到8元环孔道b酸比例高的丝光沸石分子筛,且具有在二甲醚羰基化反应中转化率高和乙酸甲酯选择性高的效果。
83.所述模板剂为n,n,n-三甲基-1-金刚烷基氢氧化铵。
84.所述模板剂n,n,n-三甲基-1-金刚烷基氢氧化铵结构式为:
[0085][0086]
可选地,所述丝光沸石分子筛骨架中si元素和al元素的摩尔比x的取值范围10≤x≤24。
[0087]
当硅铝比为10≤x≤24时,高硅丝光沸石分子筛具有更好的热稳定性,且8元环孔道b酸比例高。
[0088]
进一步优选地,所述丝光沸石分子筛骨架中si元素和al元素的摩尔比x的取值范围11≤x≤18。
[0089]
当硅铝比为11≤x≤18时,高硅丝光沸石分子筛具有二甲醚羰基化反应中转化率高,乙酸甲酯选择性高的效果。
[0090]
可选地,所述丝光沸石分子筛具有圆形纳米柱聚集成团的结构。
[0091]
可选地,所述丝光沸石分子筛x射线衍射图谱在以下位置具有特征峰:
[0092]
[0093][0094]
作为另一种实施方案,本技术还提供了一种上述任一项所述的丝光沸石分子筛的制备方法,包括以下步骤:
[0095]
将含有硅源、铝源、m源、晶种、去离子水和模板剂的初始凝胶混合物,晶化,得到所述丝光沸石分子筛;其中,所述模板剂为n,n,n-三甲基-1-金刚烷基氢氧化铵。
[0096]
可选地,所述初始凝胶混合物的制备方法包括:先将铝源、m源与去离子水混合,之后依次加入硅源、晶种、模板剂,搅拌,得到所述初始凝胶混合物。采用该顺序加料,可以得到纯相丝光沸石分子筛。
[0097]
可选地,所述初始凝胶混合物中,各组分的摩尔比为:
[0098]
sio2/al2o3=20-60;
[0099]
m2o/al2o3=1-20,其中m为碱金属离子;
[0100]
r/al2o3=1-25;
[0101]
h2o/al2o3=130-2000;
[0102]
晶种质量/投料sio2固体质量=0.1~7%。
[0103]
具体地,所述初始凝胶混合物中,sio2/al2o3的摩尔比的下限选自20、 28、36、45中的任一值;sio2/al2o3的摩尔比的上限选自28、36、45、60 中的任一值。
[0104]
m2o/al2o3的摩尔比的下限选自1.1、2.1、2.3、2.4、2.8、3、3.2、3.6、 4、4.4、4.5、5.5、6、7、7.8、8.5、10、12、15、18中的任一值;m2o/al2o3的摩尔比的上限选自2.1、2.3、2.4、2.8、3、3.2、3.6、4、4.4、4.5、5.5、6、7、7.8、8.5、10、12、20中的任一值。
[0105]
r/al2o3的摩尔比的下限选自1.2、1.6、2.4、3.5、4、4.3、4.6、4.9、5.3、5.8、6.2、6.7、7.2、7.6、7.9、8.3、8.8、11.5、13、14.5、16.0、17.5、 19、20.5、22、23.5中的任一值;r/al2o3的摩尔比的上限选自1.6、2.4、 3.5、4、4.3、4.6、4.9、5.3、5.8、6.2、6.7、7.2、7.6、7.9、8.3、8.8、11.5、 13、14.5、16.、17.5、19、20.5、22、23.5、24中的任一值。
[0106]
h2o/al2o3的摩尔比的下限选自140、180、224、250、390、450、490、 620、700、730、750、760、810、850、880、900、920、936、1200中的任一值;h2o/al2o3的摩尔比的上限选自180、224、250、390、450、490、 620、700、730、750、760、810、850、880、900、920、936、1200、1950 中的任一值。
[0107]
晶种质量/投料sio2固体质量的百分比的下限选自0.5wt%、1wt%、 2wt%、3wt%、4wt%、5wt%、6wt%中的任一值;晶种质量/投料sio2固体质量的百分比的上限选自1wt%、2wt%、3wt%、4wt%、5wt%、6wt%、7wt%中的任一值。
[0108]
优选地,所述初始凝胶混合物中,各组分的摩尔比为:
[0109]
sio2/al2o3=20-48;
[0110]
m2o/al2o3=4-12;
[0111]
r/al2o3=4-16;
[0112]
h2o/al2o3=150-1600;
[0113]
晶种质量/投料sio2固体质量=0.5~7%。
[0114]
该摩尔比可以使得丝光沸石分子筛骨架中si元素和al元素的摩尔比 x的取值范围为10≤x≤24。
[0115]
进一步优选地,所述初始凝胶混合物中,各组分的摩尔比为:
[0116]
sio2/al2o3=24-40;
[0117]
m2o/al2o3=5-12;
[0118]
r/al2o3=4-12;
[0119]
h2o/al2o3=160-1500;
[0120]
晶种质量/投料sio2固体质量=0.5~6%。
[0121]
该摩尔比可以使得丝光沸石分子筛骨架中si元素和al元素的摩尔比 x的取值范围为11≤x≤18。
[0122]
可选地,所述硅源包括硅溶胶、白炭黑、活性二氧化硅、正硅酸酯、水玻璃、偏高岭土、高岭土中的一种或任意几种的混合物。
[0123]
优选地,所述硅源为硅溶胶、白炭黑、活性二氧化硅中的至少一种。
[0124]
可选地,所述铝源包括偏铝酸钠、烷氧基铝、铝盐、偏高岭土、高岭土中的一种或任意几种的混合物。
[0125]
优选地,所述铝源为偏铝酸钠或铝盐。
[0126]
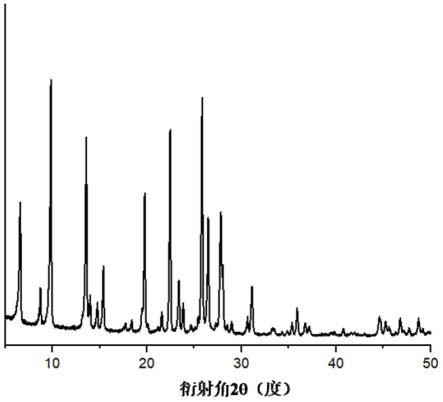
可选地,所述m源为m的氢氧化物;
[0127]
所述m的氢氧化物选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化铯中的至少一种。
[0128]
优选地,m的氢氧化物为氢氧化钠和/或氢氧化钾的至少一种。
[0129]
可选地,所述晶种选自未焙烧的丝光沸石原粉、焙烧的丝光沸石、球磨处理的丝光沸石、碱处理的丝光沸石、氟离子刻蚀处理的丝光沸石中的至少一种。
[0130]
本技术对未焙烧的丝光沸石原粉的制备方法不做严格限定,未焙烧的丝光沸石原粉可购买商业丝光沸石分子筛、或者可通过现有技术中相关文献制备、或者依据本专利合成得到的丝光沸石分子筛也可作为下次利用本专利方法合成需要的晶种。
[0131]
本技术对焙烧的丝光沸石的制备方法不做严格限定,下面介绍可能的制备方法:将上述未焙烧的丝光沸石原粉在马弗炉或管式炉中,空气气氛下(ghsv=1500-12000ml空气/g分子筛/h),450~600℃温度下焙烧2~24 h。
[0132]
本技术对球磨处理的丝光沸石的制备方法不做严格限定,下面介绍可能的制备方法:将上述未焙烧的丝光沸石原粉,利用行星式球磨机进行球磨处理1-10h。
[0133]
本技术对碱处理的丝光沸石的制备方法不做严格限定,下面介绍可能的制备方法:将上述未焙烧的丝光沸石原粉,利用0.2~1mol/l的氢氧化钠水溶液在80℃温度下处理1-24h。
[0134]
本技术对氟离子刻蚀处理的丝光沸石的制备方法不做严格限定,下面介绍可能的
制备方法:将上述未焙烧的丝光沸石原粉,利用0.1~0.5mol/l 的氟化铵水溶液在60℃温度下处理1-24h。
[0135]
可选地,所述晶化的条件为:晶化温度为120~220℃;0.5~168小时。
[0136]
可选地,晶化温度的下限独立地选自120℃、130℃、135℃、145℃、 150℃、165℃、175℃、180℃、190℃、200℃中的任一值;晶化温度的上限独立地选自130℃、135℃、145℃、150℃、165℃、175℃、180℃、 190℃、200℃、220℃中的任一值。
[0137]
晶化时间的下限独立地选自0.5h、16h、24h、32h、41h、45h、49 h、52h、58h、66h、70h、78h、90h、105h、110h中的任一值;晶化时间的上限独立地选自16h、24h、32h、41h、45h、49h、52h、58h、 66h、70h、78h、90h、105h、110h、168h中的任一值。
[0138]
优选地,晶化温度优选范围为120~200℃,进一步优选的范围为 150~180℃。
[0139]
晶化时间优选范围为10~96小时。
[0140]
可选地,所述晶化为动态晶化或者静态晶化。
[0141]
作为另一种实施方案,本技术还提供了一种催化剂,所述催化剂由上述任一项所述的丝光沸石分子筛、根据上述任一项所述制备方法得到的丝光沸石分子筛中的至少一种制备得到。
[0142]
可选地,将所述丝光沸石分子筛,经铵离子交换,再经400~700℃空气中焙烧,即可得到所述催化剂。
[0143]
作为另一种实施方案,本技术还提供了上述催化剂在在二甲醚羰基化制乙酸甲酯催化反应中的应用。
[0144]
具体地,一种二甲醚羰基化制乙酸甲酯的催化剂,所述催化剂上述丝光沸石分子筛,经铵离子交换,再经400~700℃空气中焙烧得到。
[0145]
所述二甲醚羰基化制乙酸甲酯催化反应包括:
[0146]
将含有二甲醚和一氧化碳的混合气,与所述催化剂接触、反应,得到乙酸甲酯;
[0147]
所述反应条件为:二甲醚和一氧化碳的体积比为1:5~9;
[0148]
所述混合气的空速为500~7500ml g-1
h-1
;
[0149]
反应温度为170~300℃,
[0150]
反应压力为0.5~5mpa。
[0151]
本技术中,模板剂以质量分数25%的n,n,n-三甲基-1-金刚烷基氢氧化铵水溶液的形式加入。
[0152]
本技术能产生的有益效果包括:
[0153]
1)本技术所提供的丝光沸石分子筛以n,n,n-三甲基-1-金刚烷基氢氧化铵作为模板剂,制备得到的催化剂8元环孔道b酸比例高,具有更好的催化性能。
[0154]
2)本技术所提供的催化剂,8元环孔道中的b酸中心数量在丝光沸石分子筛总b酸中心数量中的占比为80%~92%,在二甲醚羰基化制乙酸甲酯催化反应中表现出优异的催化性能。
[0155]
3)本技术所提供的二甲醚羰基化制乙酸甲酯的方法,二甲醚的转化率可达到92%以上,乙酸甲酯选择性可达到99%以上。
附图说明
[0156][0157]
图1为实施例1中的样品的x射线衍射图谱;
[0158]
图2为实施例1中的样品的扫描电子显微镜图;
[0159]
图3为实施例1中的样品热重谱图;
[0160]
图4为对比例1中的产物的x射线衍射图谱;
[0161]
图5为对比例2中的产物的x射线衍射图谱;
[0162]
图6为对比例3中的产物的扫描电子显微镜图;
[0163]
图7为对比例4中的产物的x射线衍射图谱;
[0164]
图8为实施例22所制催化剂的红外谱图;
[0165]
图9为实施例22所制催化剂在二甲醚羰基化反应中的催化性能图;
[0166]
图10为对比例5所制催化剂的红外谱图;
[0167]
图11为对比例6所制催化剂的红外谱图。
具体实施方式
[0168]
下面结合实施例详述本技术,但本技术并不局限于这些实施例。
[0169]
如无特别说明,本技术的实施例中的原料均通过商业途径购买。
[0170]
下面介绍可能的实施方式:
[0171]
本技术的目的在于提供一种8元环孔道b酸比例高的丝光沸石分子筛,该分子筛的无水化学组成可表示为:nr
·
mm
·
(si
x
al)o
2x+2
,r为模板剂分子,为n,n,n-三甲基-1-金刚烷基氢氧化铵。n为每摩尔(si
x
al)o
2x+2
中模板剂r的摩尔数n=0.05-0.85;m为碱金属离子,m为每摩尔(si
x
al)o
2x+2
中碱金属离子的摩尔数,m=0.15-0.95;x为丝光沸石分子筛骨架中si元素和al元素的摩尔比,x=10-30。
[0172]
所述的分子筛x射线衍射图谱在以下位置具有特征峰:
[0173][0174]
所述分子筛骨架中硅元素与铝元素的摩尔比x在10-30;
[0175]
进一步地,所述分子筛骨架中硅元素与铝元素的摩尔比x优选10-24;
[0176]
更进一步地,所述分子筛骨架中硅元素与铝元素的摩尔比x优选11-18。
[0177]
本技术的另一目的在于提供一种上述丝光沸石分子筛的合成方法。
[0178]
本发明所要解决的技术问题是直接以n,n,n-三甲基-1-金刚烷基氢氧化铵有机化合物作为模板剂,以常规丝光沸石分子筛合成所用的硅源、铝源、碱源为原料,在水热合成条件下合成制备纯相丝光沸石分子筛。本发明提供该丝光沸石分子筛的水热合成方法。
[0179]
所述的丝光沸石分子筛的合成方法,该方法的合成步骤如下:
[0180]
a)硅源、铝源、碱金属m的氢氧化物、晶种、去离子水和模板剂r 混合,形成具有如下摩尔配比的初始凝胶混合物:
[0181]
sio2/al2o3=20-60;
[0182]
m2o/sio2=0.1-0.35,其中m为碱金属;
[0183]
r/sio2=0.05-0.60;
[0184]
h2o/sio2=6-40;
[0185]
晶种质量/投料sio2固体质量=0.1~7%;
[0186]
b)将步骤a)处理后得到的初始凝胶混合物于装入水热合成釜,密闭,升温到120~220℃下晶化0.5~168小时;
[0187]
c)待步骤b)晶化完成后,固体产物经分离、洗涤、干燥后即得所述的丝光沸石分子筛。
[0188]
其中,所述步骤a)中的硅源为硅溶胶、活性二氧化硅、正硅酸酯、水玻璃、偏高岭土中的一种或任意几种的混合物;优选地,所述硅源为硅溶胶、白炭黑、活性二氧化硅中的至少一种。
[0189]
所述步骤a)中的铝源为偏铝酸钠、烷氧基铝、铝盐、偏高岭土中的一种或任意几种的混合物;优选地,所述铝源为偏铝酸钠或铝盐。
[0190]
所述步骤a)中的碱金属m的氢氧化物为氢氧化锂、氢氧化钠、氢氧化钾、氢氧化铯中的至少一种;优选地,所述碱金属的氢氧化物为氢氧化钠和氢氧化钾的至少一种。
[0191]
所述步骤a)中的晶种为未焙烧的丝光沸石原粉、焙烧的丝光沸石、球磨处理的丝光沸石、碱处理的丝光沸石、氟离子刻蚀处理的丝光沸石中的至少一种。优选地,所述晶种为未焙烧的丝光沸石原粉和碱处理的丝光沸石中的至少一种。
[0192]
所述步骤a)初始凝胶混合物中sio2/al2o3的摩尔比优选范围为20-48,进一步的优选范围为24-40。
[0193]
所述步骤a)初始凝胶混合物中m2o/sio2的摩尔比优选范围为 0.1-0.35,进一步的优选范围为0.12-0.24。
[0194]
所述步骤a)初始凝胶混合物中r/sio2的摩尔比优选范围为0.05-0.60。
[0195]
所述步骤a)初始凝胶混合物中h2o/sio2的摩尔比优选范围为6-40,进一步的优选范围为8-20。
[0196]
所述步骤b)中的晶化温度优选范围为120~200℃,进一步的优选范围为150~180℃。
[0197]
所述步骤b)中的晶化时间优选范围为10~96小时。
[0198]
所述步骤b)中的晶化过程可以在静态进行,也可以在动态进行。
[0199]
本技术的又一目的在于还提供了一种二甲醚羰基化制乙酸甲酯催化剂,该催化剂可以应用于二甲醚羰基化制乙酸甲酯催化反应,并表现出优异的催化性能。
[0200]
所述催化剂由上述的丝光沸石分子筛、根据上述任一方法合成的丝光沸石分子筛中的至少一种,经铵离子交换、再经400~700℃空气中焙烧得到。
[0201]
本技术的实施例中分析方法如下:
[0202]
x射线粉末衍射物相分析(xrd)测试采用荷兰帕纳科(panalytical) 公司的x’pertprox射线衍射仪,cu靶,kα辐射源(λ=0.15418nm),电压40kv,电流40ma。
[0203]
实施例中体相元素组成测定采用philips公司的magix2424x型射线荧光分析仪(xrf)测定。
[0204]
扫描电子显微镜(sem)测试所采用仪器为hitachisu8020场发射扫描电镜,加速电压为2kv。
[0205]
红外透射光谱(ftir)实验在真空系统中进行,样品450℃脱水处理,室温采谱。
[0206]
采用美国ta公司的sdtq600热分析仪对样品进行程序升温条件下的重量变化和热流分析。空气气氛,流速100ml/min。
[0207]
气体样品分析采用美国安捷伦(agilent)公司6890gc型气相色谱仪进行在线分析,色谱柱为安捷伦(agilent)公司poraplotq毛细柱。
[0208]
二甲醚的转化率=[(混合气中的二甲醚碳摩尔数)-(产物中的二甲醚碳摩尔数)]/(混合气中的二甲醚碳摩尔数)*100%
[0209]
乙酸甲酯的选择性=(2/3)*(产物中的乙酸甲酯碳摩尔数)/[(混合气中的二甲醚碳摩尔数)-(产物中的二甲醚碳摩尔数)]*100%
[0210]
未焙烧的丝光沸石原粉晶种的制备方法:参见文献(acs catalysis, 2020,10,3372-3380)。
[0211]
焙烧的丝光沸石晶种的制备方法:将利用上述文献方法制备的丝光沸石原粉分子筛,在马弗炉或管式炉中,空气气氛下(ghsv=1500ml空气 /g分子筛/h),500℃温度下焙烧12h。
[0212]
球磨处理的丝光沸石晶种的制备方法:将上述未焙烧的丝光沸石原粉,利用行星式球磨机进行球磨处理5h。
[0213]
碱处理的丝光沸石晶种的制备方法:将上述未焙烧的丝光沸石原粉,利用0.5mol/l的氢氧化钠水溶液在80℃温度下处理12h。
[0214]
氟离子刻蚀处理的丝光沸石晶种的制备方法:将上述未焙烧的丝光沸石原粉,利用0.3mol/l的氟化铵水溶液在60℃温度下处理10h。
[0215]
实施例1
[0216]
初始凝胶中各原料的摩尔比例和晶化条件见表2。首先将0.8g偏铝酸钠和1.48g氢氧化钠固体溶解于18.87g去离子水中混合均匀后,将32.92 g硅溶胶(27.3wt%)在搅拌的状态下,缓慢滴加到上述溶液。继续向该混合物中一次加入0.5g未焙烧的丝光沸石原粉晶种和18.39g n,n,n-三甲基-1-金刚烷基氢氧化铵,之后将形成的初始凝胶继续在室温下继续搅拌直至均匀。将上述凝胶转移放入带聚四氟乙烯内衬的不锈钢反应釜中,升温至185℃在动态条件下晶化32h,所得固体产物经离心分离,用去离子水洗涤至中性,在110℃下空气中干燥,得到原粉(即本技术中的丝光沸石分子筛)。
[0217]
对实施例1产品做xrd分析,结果表明合成产物具有丝光沸石分子筛的特征(xrd谱图见图1)。对实施例1所得到的样品进行扫描电镜表征。样品的扫描电子显微镜图如图2所示,样品呈现圆形纳米柱聚集成团的结构,圆形纳米柱的直径约为200~300nm。
[0218]
采用xrf分析分子筛晶体的体相硅铝组成,结果列于表2。实施例1 样品的体相si/al摩尔比为15.2。
[0219]
对实施例1原粉样品进行热重分析(热重谱图见图3),显示有机失重占分子筛干基质量的8%。将热重分析和xrf测定的无机元素组成归一化,得到实施例1的丝光沸石分子筛的无水化学组成为 0.61r
·
0.39na
·
(si
15.2
al)o
32.4
,其中r为n,n,n-三甲基-1-金刚烷基氢氧化铵。
[0220]
对实施例1丝光沸石原粉样品进行
13
c mas nmr分析,仅发现归属于n,n,n-三甲基-1-金刚烷基氢氧化铵的特征碳共振峰,说明n,n, n-三甲基-1-金刚烷基氢氧化铵在晶化过程保持结构完整,且作为模板剂被包裹到所得丝光沸石分子筛的孔道之中。
[0221]
图1特征峰对应的2θ如表1所示,表明合成产物具有丝光沸石分子筛的特征。
[0222]
表1图1特征峰对应的2θ
[0223][0224][0225]
实施例2-19
[0226]
实施例2-19的具体配料比例和晶化条件见表2,具体配料过程同实施例1。对实施例2-19合成得到原粉样品做xrd分析,产品的x-射线衍射谱图具有图1的特征,即峰位置和形状基本相同,依合成条件的变化衍射峰的相对峰强对在
±
10%范围内波动,证明合成产物均为丝光沸石分子筛。
[0227]
采用xrf分析实施例2-19分子筛的体相硅铝元素组成,相应的体相硅铝组成列于表2。丝光沸石分子筛的无水化学组成见表3所示。
[0228]
表2分子筛初始凝胶配料、晶化条件及产物体相和表面硅铝元素组成*
[0229][0230]
注
*
:硅源:a硅溶胶、b活性二氧化硅、c正硅酸酯、d水玻璃、e偏高岭土。
[0231]
铝源:f偏铝酸钠、g烷氧基铝、h铝盐、i偏高岭土。
[0232]
模板剂:n,n,n-三甲基-1-金刚烷基氢氧化铵。
[0233]
晶种:i未焙烧的丝光沸石原粉、
ii
焙烧的丝光沸石、
iii
球磨处理的丝光沸石、
iv
碱处理的丝光沸石、v氟离子刻蚀处理的丝光沸石。
[0234]
晶化条件:α动态晶化、β静态晶化。
[0235]
晶种的添加量为晶种质量/投料sio2固体质量。
[0236]
表3丝光沸石分子筛的无水化学组成
[0237]
实施例无水化学组成20.56r
·
0.44na
·
(si
19.8
al)o
41.6
30.61r
·
0.39na
·
(si
11.2
al)o
24.4
40.65r
·
0.35na
·
(si
12.5
al)o
27
50.41r
·
0.59na
·
(si
13.1
al)o
28.2
60.51r
·
0.49na
·
(si
21.3
al)o
44.6
70.75r
·
0.25na
·
(si
26.8
al)o
55.6
80.79r
·
0.21na
·
(si
16.3
al)o
34.6
90.75r
·
0.25na
·
(si
15.2
al)o
32.4
100.42r
·
0.58na
·
(si
14.7
al)o
31.4
110.28r
·
0.72na
·
(si
14.8
al)o
31.6
120.56r
·
0.44na
·
(si
11.9
al)o
25.8
130.49r
·
0.51na
·
(si
12.6
al)o
27.2
140.46r
·
0.54na
·
(si
11.8
al)o
25.6
150.72r
·
0.28na
·
(si
13.2
al)o
28.4
160.66r
·
0.34na
·
(si
14.1
al)o
30.2
170.49r
·
0.51na
·
(si
14.8
al)o
31.6
180.59r
·
0.41na
·
(si
13.3
al)o
28.6
190.57r
·
0.43na
·
(si
12.8
al)o
27.6
[0238]
实施例20
[0239]
分别取实施例1-19的合成样品3g,放入塑料烧杯中,于冰水浴条件下加入3ml40%的氢氟酸溶液溶解分子筛骨架,然后加入15ml三氯甲烷溶解其中的有机物。将有机物用gc-ms分析组成显示其中所含的有机物均为对应合成过程中采用的模板剂r。
[0240]
实施例21
[0241]
配制摩尔配比为40sio2:1al2o3:6.2k2o:840h2o:5.8r:5.5wt%晶种的初始凝胶,其中r为n,n,n-三甲基-1-金刚烷基氢氧化铵。将铝源更改为无水氯化铝,碱源更改为90wt%的氢氧化钾,其他原料同实施例1。具体配料过程和晶化条件同实施例1。产品做xrd分析,产品的x-射线衍射谱图具有图1的特征,即峰位置和形状基本相同,证明合成产物为丝光沸石分子筛。
[0242]
采用xrf分析分子筛晶体的体相硅铝组成,实施例21样品的体相 si/al摩尔比为14.5。
[0243]
对比例1
[0244]
除不添加有机模板剂之外,其他配料比例和配料过程,以及晶化条件同实施例1。所得产物经xrd鉴定为丝光沸石和zsm-5的混合物。相应 xrd图谱见图4。
[0245]
对比例2
[0246]
除不添加有机模板剂之外,其他配料比例和配料过程,以及晶化条件同实施例21。所得产物经xrd鉴定为丝光沸石和镁碱沸石的混合物。相应xrd谱图见图5。
[0247]
对比例3
[0248]
配制摩尔配比为40sio2:1al2o3:6.2na2o:840h2o:5.8teaoh:5.5wt%晶种的初始凝胶。仅将模板剂更改为25wt%的四乙基氢氧化铵(teaoh) 水溶液,其他原料同实施例1。具体配料过程和晶化条件同实施例1。产品做xrd分析,产品的x-射线衍射谱图具有图1的特征,即峰位置和形状基本相同,证明合成产物为丝光沸石分子筛。
[0249]
样品的扫描电子显微镜图如图6所示,由图6可以看出,当模板剂改为teaoh时,合成的分子筛形貌为尺寸在2~3微米的块状晶体,与本技术的分子筛形貌差别非常大,可见,本技术提供的模板剂对形成圆形纳米柱聚集成团的结构的形貌至关重要。
[0250]
对比例4
[0251]
配制摩尔配比为40sio2:1al2o3:6.2na2o:840h2o:5.8r的初始凝胶,其中 r为n,n,n-三甲基-1-金刚烷基氢氧化铵。除不再添加晶种外,其他原料同实施例1。具体配料过程和晶化条件同实施例1。产品做xrd分析,所得产物为丝光沸石和无定形氧化硅的混合物。相应xrd图谱见图7。
[0252]
实施例22
[0253]
将实施例1得到的样品于600℃通入干燥空气焙烧4h,使用1mol/l 的nh4no3溶液在80℃下搅拌1h进行离子交换(固液比为1:10)去除钠离子,此过程重复三次,水洗干燥后在550℃空气中焙烧4h,压片、破碎为粒度为40~60目的催化剂颗粒,记为1#催化剂样品,8元环孔道b酸占总b酸量的比例为85%(红外谱图分峰结果见图8)。
[0254]
称取1.0g 1#催化剂在固定床反应器中进行二甲醚(简写为dme) 羰基化反应评价。反应开始时在400℃下通氮气活化1h,然后降温至 300℃。以30ml/min的气体流速携带吡啶通入反应器,处理1h,之后氮气吹扫1h(30ml/min)。最后降温至200℃进行反应。混合气 (dme/co/n2=2/14/84,体积比),气体空速为3000ml g-1
h-1
(stp),反应压力为2.0mpa。反应达到平衡(约6h)之后dme的转化率92%,乙酸甲酯选择性大于99.9%。相应二甲醚羰基化反应性能图见图9。
[0255]
对比例5
[0256]
除不添加有机模板剂和在静态条件下晶化之外,其他配料比例和配料过程,以及晶化条件同实施例1。所得产物经xrd鉴定为mor,产物硅铝比(sio2/al2o3)为18.2。
[0257]
将对比例5中的样品经nh4no3离子交换去除钠离子(同实施例22 中的处理方法),550℃空气中焙烧4h后,压片、破碎至40~60目,记为2#催化剂样品,8元环孔道b酸占总b酸量的比例为52%(红外谱图分峰结果见图10)。称取1.0g 2#催化剂在固定床反应器中进行二甲醚 (简写为dme)羰基化反应评价。反应开始时在400℃下通氮气活化1 h,然后降温至300℃。以30ml/min的气体流速携带吡啶通入反应器,处理1h,之后氮气吹扫1h(30ml/min)。最后降温至200℃进行反应。混合气(dme/co/n2=2/14/84,体积比),气体空速为3000ml g-1
h-1
(stp),反应压力为2.0mpa。经过6h诱导期后,取样得到dme的转化率和产物中乙酸甲酯的选择性。二甲醚的转化率52%,乙酸甲酯选择性99%,二甲醚转化率明显低于实施例22。可见,本技术提供的模板剂对合成具有高8元环孔道b酸比例且二甲醚羰基化反应活性优异的丝光沸石样品至关重要。
[0258]
对比例6
[0259]
将对比例3制备的丝光沸石分子筛样品于600℃通入干燥空气焙烧4 h,nh4no3离子交换去除钠离子(同实施例22中的处理方法),550℃空气中焙烧4h后,压片、破碎至40~60目,所得产物记为3#催化剂样品,经xrd鉴定为mor,8元环孔道b酸占总b酸量的比例为47%(红外谱图分峰结果见图11)。
[0260]
称取1.0g 3#催化剂在固定床反应器中进行二甲醚(简写为dme) 羰基化反应评价。反应开始时在400℃下通氮气活化1h,然后降温至 300℃。以30ml/min的气体流速携带吡啶通入反应器,处理1h,之后氮气吹扫1h(30ml/min)。最后降温至200℃进行反应。混合气 (dme/co/n2=2/14/84,体积比),气体空速为3000ml g-1
h-1
(stp),反应压力为2.0mpa。经过6h诱导期后,取样得到dme的转化率和产物中乙酸甲酯的选择性。二甲醚转化率下降到只有40%,乙酸甲酯选择性99%,二甲醚转化率明显低于实施例22。可见,本技术提供的模板剂对合成具有高8元环孔道b酸比例且二甲醚羰基化反应活性优异的丝光沸石样品至关重要。
[0261]
以上所述,仅是本技术的几个实施例,并非对本技术做任何形式的限制,虽然本技术以较佳实施例揭示如上,然而并非用以限制本技术,任何熟悉本专业的技术人员,在不脱离本技术技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1