一种阴离子原位掺杂高镍三元正极材料的制备方法
1.本发明涉及锂电高镍前驱体及正极材料技术领域,具体涉及一种阴离子原位掺杂高镍三元前驱体及其正极材料的制备方法。
背景技术:
2.随着新能源交通工具的快速发展,锂离子电池作为其重要的动力和能量源而被大规模生产并广泛投入商业应用。伴随电动汽车对续航里程、成本及安全性要求的不断提升,动力锂离子电池的研发制造必须将能量密度、成本及材料稳定性提升到一个新的高度。在锂离子电池的各个组成部分中,正极材料是决定电池综合性能最核心部件之一,也是锂电性能提升的瓶颈所在。为满足高能量密度及低成本的发展趋势,高镍层状三元材料lini
x
coymn
1-x-y
o2(x≥0.6)由于ni含量高具有放电容量高(170mah/g以上)、成本低、综合性能可提升空间大等优势成为动力锂离子电池首先正极材料之一。
3.但是随着ni含量的升高,高镍材料的结构稳定性及安全性也逐渐恶化,在材料制备的过程中易于发生li/ni混排严重、表面残碱量高、难以形成化学计量比产物等行业难题,从而导致材料可逆容量衰减过快、充放电过程电荷转移阻抗增加、循环过程中材料与电解液界面副反应加剧(例如,充电态时,高价ni
4+
易于与电解液中的hf反应造成金属溶解),给高镍材料的大规模推广使用带来了极大的安全隐患。
4.为解决上述难题,行之有效的一个方法就是向材料的体相结构中掺杂一些金属或非金属元素,使其在材料中起稳定结构或阻隔电极副反应的作用,保障材料在长循环过程中结构保持相对完整,提升电极/电解液之间副反应发生的能垒。而金属离子的掺杂通常能起到稳定材料结构、增强导电性的作用,但在抑制电极/电解液副反应、晶格氧的析出方面效果不明显;而阴离子掺杂恰好弥补这一不足,且阴离子掺杂能够增强其与材料过渡金属之间的键合力,有效提升材料的结构稳定性和长循环特性。但阴离子掺杂一般采用在前驱体固相烧结中混入掺杂元素或将正极材料与掺杂物料混合二次烧结,无疑增加了处理工序及成本,且掺杂元素通过固相扩散易于造成分布不均,使高镍材料出现性能不稳定甚至恶化。例如;中国专利cn111377487a公开了一种al、f共掺杂高镍三元正极材料的制备方法,该方法是将前驱体、锂源、al添加剂及氟添加剂经过球磨、煅烧后实现元素的掺杂,这样的掺杂方式容易使球磨过程中前驱体颗粒破碎,从而导致元素掺杂的均匀性和准确性得不到保证。
技术实现要素:
5.针对高镍材料长循环过程中易于与电解液发生副反应导致材料过渡金属溶解、晶格氧缺失,进而使高镍材料循环性能恶化,本发明提供一种阴离子f-原位掺杂高镍三元前驱体及其正极材料的制备方法。本发明通过液相共沉淀法使金属盐原料液中的阴离子f-在前驱体形核和生长过程中均匀扩散至高镍球形前驱体内部,所得前驱体结合固相锂化煅烧获得f均匀掺杂的高镍正极材料,f的掺杂使高镍层状材料的结构更加稳定、循环性能突出。
为实现上述目标,所采用的技术方案为:本发明阴离子f-原位掺杂高镍前驱体及其正极材料的制备过程包括:(1)金属盐原料液的配制;(2)镍钴锰共沉淀反应制备高镍前驱体ni
x
coymn
1-x-y
(oh)2(0.6≤x<1);(3)前驱体混锂富氧煅烧合成正极材料;其中,f-是在金属盐原料液中配入,在前驱体共沉淀反应中原位掺杂进入前驱体颗粒中。
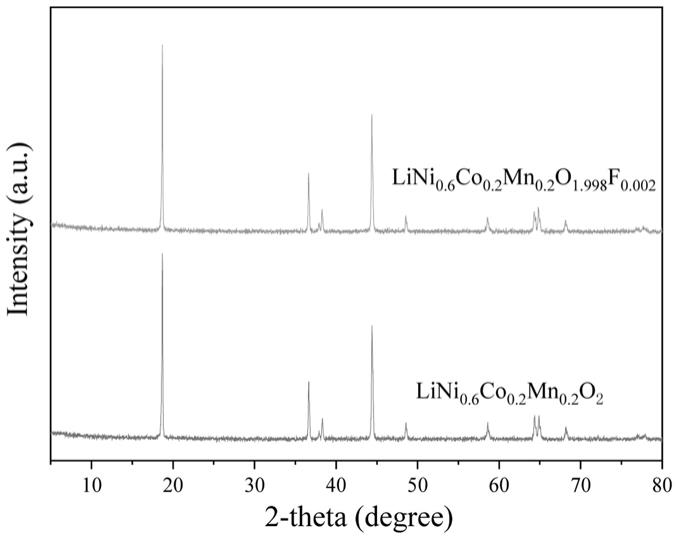
6.进一步的,所述步骤(1)原料溶液配制包括:含f-镍钴锰金属可溶性盐溶液ⅰ、氨水作为共沉淀反应络合剂ⅱ、碱液作为沉淀剂ⅲ(调控釜内料液ph的),反应进料前还应配制一定浓度的氨水作为反应底液ⅳ。
7.进一步的,所述步骤(1)含f-镍钴锰金属可溶性盐溶液ⅰ配制包括:ni、co、mn元素比按材料设计比例配制,将f源与各金属盐称取好后置于储罐内,加40-50℃的水快速溶解(采用热水快速溶解并通过热水降低溶液中的氧含量,而后转移到容量瓶中冷却后定容),使ni、co、mn金属总浓度在1.5-2.5mol/l,f在金属盐溶液中浓度为0.1-1.9g/l。
8.或直接采用废旧锂电正极材料的回收浸出液,将浸出液中的各组分浓度调配至合适比例后作为金属盐溶液,具体是将废液中的各组分浓度调配至ni、co、mn金属总浓度在1.5-2.5mol/l,f在金属盐溶液中浓度为0.1-1.9g/l;从而大大缩减成本。
9.进一步的,所述步骤(1)络合剂氨水ⅱ的配制包括:氨水络合剂的浓度范围应参考金属盐溶液的浓度、进料速度及氨水的进料速度,使金属盐与氨的进料摩尔比始终维持在nh3:tm(tm=ni+co+mn)=0.8-1.4范围内(tm=ni、co、mn);进一步的,所述步骤(1)碱液沉淀剂ⅲ的配制包括:碱液配制的浓度范围为4.0-8.0mol/l。
10.进一步的,所述步骤(1)氨水底液ⅳ的配制包括:底液体积为加料完毕后反应料液总体积的1/4-1/2,底液氨浓度与进料氨在三股进料液中的浓度基本一致。
11.步骤(1)所述方法中,镍钴锰的可溶性金属盐可选用镍钴锰的金属硫酸盐、氯盐及硝酸盐中的一种或多种;碱液可选用lioh、naoh、koh等可溶性强碱中的一种或多种;f源为可溶性naf、kf或nh4f中的一种或多种。
12.本发明中,步骤(2)镍钴锰共沉淀反应制备高镍前驱体ni
x
coymn
1-x-y
(oh)2(0.6≤x<1)包括:前驱体制备反应开启后,通过控制碱液泵的流速维持反应料液的ph在10.5-11.5范围内;反应釜料液温度恒定在50-60
°
c内;镍钴锰金属盐溶液的流速为0.7-10.0ml/min;进料时间为6-50h;搅拌速度500-1000rpm/min。
13.进一步的,所述步骤(2)前驱体反应进料结束后,继续维持搅拌和加热6-12h,而后静置3-6h。即,碱液泵的流速维持反应料液的ph在10.5-11.5范围内,反应釜料液温度恒定在50-60
°
c内;并在500-1000 rpm下维持进料6-50h进行共沉淀反应,共沉淀反应进料结束后,继续维持搅拌和加热6-12h,而后静置3-6h。
14.进一步的,所述步骤(2)前驱体反应结束后,料液通过静置,倒掉上清液,反复用去离子水洗涤5遍以上,以去除前驱体中夹杂的na
+
、so
42-等杂质;前驱体滤饼在真空烘箱中100-120
°
c干燥24h左右,以去除夹杂的水分。
15.本发明中,步骤(3)前驱体混锂富氧煅烧合成正极材料包含:将前驱体与锂源用乙醇分散剂混合研磨30min以上直至乙醇完全挥发,将混锂前驱体粉末至于匣钵中压实,将匣钵至于管式炉中进行富氧烧结,待自然冷却后将正极粉末研磨过筛,最后用样品袋密封干
燥保存。
16.进一步的,步骤(3)所述锂源为lioh微米级粉末;匣钵采用刚玉氧化铝坩埚;烧结温度分为两段,第一段以5-10
°
c/min升温至400-500
°
c保温4-5h,第二段以5-10
°
c/min升温至720-900℃保温10-12h(ni含量越高温度越低);优选方案中,升温速率为5
°
c/min,第一段升温至500℃,第二段升温至850℃;粉末过筛采用200目筛网。
17.本发明的优点及有益效果为:(1)向高镍材料中掺杂电负性较o
2-更强的阴离子f-,由于tm-f键的键能强于tm-o键,使f掺杂增强了高镍材料过渡金属骨架结构的稳定性;同时,由于f-是均匀迁移到前驱体颗粒内部,从而有效避免了颗粒表面f过量,而中心位置f短缺带来的掺杂不均匀,进而降低由掺杂不均带来的材料充放电过程中一次晶粒之间的内部应力,减少颗粒微裂纹的产生,提升材料的长循环稳定性;(2)由于tm-f强于tm-o,使循环过程中由电解液分解的少量hf与tm过渡金属反应的能垒增高,从而增强了电极/电解液的界面稳定性;(3)由于废旧锂电池湿法回收过程中得到的含ni、co、mn的有价金属溶液中不可避免的包含有f-,通过各组分的调配满足材料制备要求,可作为本发明的含f金属盐原料液,极大程度地降低了生产成本,实现了金属资源的循环利用。
18.(4)由于三元前驱体锂化烧结反应是氧化反应,在煅烧过程前驱体过渡金属ni、co、mn在有氧条件下氧化为相应的高价态过渡金属并与锂源相互融合发生反应最终生成层状的锂过渡金属氧化物,因此反应必须在有氧条件下进行,其反应方程式可表示为:m(oh)2+lioh
·
h2o+0.25o2=limo2+2.5h2o(锂源为氢氧化锂)m(oh)2+0.5li2co3+0.25o2=limo2+0.5co2+h2o(锂源为碳酸锂)(5)如果在无氧条件下(如惰性气氛ar气)进行前驱体的锂化烧结实验,反应产物为相应的nio、coo、mno等低价金属氧化物,而非三元层状结构的正极材料,所得材料很可能不具有电化学性能。
附图说明
19.图1 为实施例1未掺氟原样lini
0.6
co
0.2
mn
0.2
o2及掺氟lini
0.6
co
0.2
mn
0.2o1.998f0.002
材料的xrd图。
20.图2 (a)、(c)分别为实施例1未掺氟lini
0.6
co
0.2
mn
0.2
o2材料的前驱体和其正极材料的sem图片;(b)、(d)分别为实施例1掺氟lini
0.6
co
0.2
mn
0.2o1.998f0.002
材料的前驱体和其正极材料的sem图片。
21.图3 为实施例1管式炉富氧条件下煅烧的未掺氟原样lini
0.6
co
0.2
mn
0.2
o2及掺氟lini
0.6
co
0.2
mn
0.2o1.998f0.002
材料在1c下,2.8-4.3v电压区间内的放电容量衰减曲线。
22.图4 为实施例1马弗炉空气条件下煅烧的未掺氟原样lini
0.6
co
0.2
mn
0.2
o2及掺氟lini
0.6
co
0.2
mn
0.2o1.998f0.002
材料在1c下,2.8-4.3v电压区间内的放电容量衰减曲线。
23.图5为实施例1金属盐溶液中引入不同f-浓度梯度下合成的正极材料在1c下,2.8-4.3v电压区间内的放电容量衰减曲线。
具体实施方式
24.以下结合实施例对本发明中一种阴离子原位掺杂高镍三元前驱体及其正极材料的制备方法作进一步说明,而不会形成对本发明的限制。
25.实施例1:f-掺杂的高镍lini
0.6
co
0.2
mn
0.2
o2材料的制备(1)反应原料液的配制1)按化学计量比ni:co:mn=6:2:2准确称取可溶性的niso4·
6h2o、coso4·
7h2o及mnso4·
h2o盐置于烧杯中,再加入一定量的naf作为f源(不掺f原样不添加f源,其他操作步骤均相同),加入一定量热纯水(50℃)搅拌并超声溶解,待冷却至常温后转移至容量瓶中定容获得金属盐溶液ⅰ,其中,镍钴锰的总金属浓度为2.0mol/l,f-在金属盐溶液中的浓度分别为0、0.95g/l、1.9g/l、3.8g/l、5.5g/l;2)用移液管准确量取一定体积的浓氨水于容量瓶中,加入纯水定容获得2.2mol/l氨水络合剂ⅱ;3)快速称取一定量的可溶性碱氢氧化钠于烧杯内,加入冷的纯水并在冷水浴中超声溶解,而后快速转移到容量瓶中定容,获得碱液沉淀剂ⅲ,其浓度为4.0mol/l;4)氨水底液的配制同2)相同,底液体积为加料完毕后反应料液总体积的1/2,底液氨浓度为0.7mol/l;(2)液相共沉淀反应制备掺f高镍前驱体1)实验开始前先对ph计进行两点校准,反应前先将氨水底液倒入反应釜中,底液体积为加料完毕后反应料液总体积的1/2,通过水浴锅向反应釜夹层泵入循环水加热釜内温度,与此同时向釜内持续通入氮气排尽空气(氮气通入液面以下),待釜内温度升至反应温度后,通过三台加料泵将三股料液泵至加料口末端后停止(保证三股料液同时进入釜内);2)设置三台进料泵的流量,其中泵1用来抽送金属盐溶液,其流量控制在0.7l/min,进料时间为6h左右;泵2、3分别用来抽送氨水和碱液,其流量设定根据泵1金属盐进料速度换算而来,使金属盐与氨水的进料摩尔比始终维持在nh3:tm(tm=ni+co+mn)=1.2;准备工序完成后,同时开启三台进料泵并开始计时,反应过程中,控制釜内温度为55
°
c,维持料液ph恒定在10.6,搅拌速度为800rpm;加料结束后,继续维持加热搅拌8h,而后静置4h后放料收集前驱体料液,前驱体洗涤5遍后真空过滤,将滤饼转移至真空干燥箱100
°
c干燥24h,研磨后将前驱体装入密封袋干燥保存得到前驱体。
26.根据制备前驱体中,f-在金属盐溶液中的浓度分别为0、0.95g/l、1.9g/l、3.8g/l、5.5g/l分别得到前驱体1、前驱体2、前驱体3、前驱体4、前驱体5;(3)前驱体混锂煅烧制备掺f正极材料lini0.6co0.2mn0.2o2将上述不同前驱体分别与微米级lioh
·
h2o用乙醇作分散剂均匀混合研磨(li:tm=1.03),而后装入匣钵中置于管式炉内通入氧气煅烧,升温速率为5
°
c/min,煅烧分为两段,第一段500
°
c保温5h,第二段850
°
c保温12h,而后自然冷却至80
°
c取出后研磨过200目筛,分别得到原样lini0.6co0.2mn0.2o2及氟掺杂的lini0.6co0.2mn0.2o2fx高镍正极材料,其中,根据氟离子选择电极法,当金属盐溶液中f-浓度为1.9g/l时,经过前驱体共沉淀及固相煅烧制备的掺f样品中f的含量经换算用化学式可表示为
lini0.6co0.2mn0.2o1.998f0.002。
27.(4)材料性能测试将步骤(3)得到的正极材料、pvdf粘结剂、乙炔黑导电碳按质量比8:1:1称取后混合研磨,然后加入nmp溶剂搅拌混合3h得到正极浆料,将正极浆料均匀涂附于铝箔上110
°
c烘干24h获得正极片,正极片通过冲压机剪切成直径12mm的圆片,采用金属锂片作负极,在氩气手套箱中组装成扣式电池,在蓝电测试系统上进行电池充放电性能测试。图3为1c(1c=180mah/g)电流密度下,2.8-4.3v电压区间内未掺f原样及金属盐溶液中f-浓度为1.9g/l时合成正极材料(即lini0.6co0.2mn0.2o1.998f0.002)的放电容量衰减曲线(氧气气氛煅烧),未掺f材料的首圈放电容量为156.3mah/g,100圈后的放电容量为121.4mah/g,容量保持率为77.7%;而f-浓度为1.9g/l时样品的首圈放电容量为154.9mah/g,100圈循环后的放电容量为139.6mah/g,容量保持率为90.1%,可见f的掺杂虽然造成了初始容量的降低,但其循环稳定性得以明显改善(注:容量衰减曲线初始阶段出现缓慢上升或波动是由于电池循环初期的活化效应,下同)。
28.图3为金属盐溶液中不同f-浓度梯度下(f-浓度分别为0、0.95g/l、1.9g/l、3.8g/l、5.5g/l)合成的正极材料的放电容量衰减曲线,可以看出,随着金属盐溶液中f-浓度的升高,材料的初始放电容量逐步下降,首圈放电容量分别为156.3mah/g(f浓度为0)、157.3mah/g(f浓度为0.95g/l)、154.9mah/g(f浓度为1.9g/l)、148.4mah/g(f浓度为3.8g/l)、141.8mah/g(f浓度为5.5g/l),当f-浓度从0增大到1.9g/l时,材料的循环稳定性逐步增加,而进一步增大金属盐溶液中的f-浓度,不仅会导致材料容量的损失,其循环稳定性由于阳离子混排过于严重也将逐步恶化,因此金属盐溶液中可容纳的最佳f-浓度范围为0-1.9g/l。
29.实施例2方法、步骤同实施例1,以154.9mah/g(f浓度为1.9g/l)为例,仅将富氧条件为充空气烧结,烧结的温度等条件同实施例1。图4为图3将烧结气氛改为空气时合成的相应未掺f原样及掺f材料的放电容量衰减曲线,可以看出将烧结气氛改为空气,材料的循环性能均有所下降,其中未掺f原样的初始放电容量为156.4mah/g,100圈后放电容量衰减至100mah/g;而掺f(f浓度为1.9g/l)材料的首圈放电容量为152.8mah/g,100圈后的容量降至118.4mah/g,可见掺f样品的初始容量虽然有所降低,但循环性能优于未掺f原样。
30.实施例3方法、步骤同实施例1,以154.9mah/g(f浓度为1.9g/l)为例,仅将富氧条件为充氮气烧结,烧结的温度等条件同实施例1,得到的产品不具有电化学性能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1